
Галактоземия (galactosaemia; греч. gala, galaktos молоко + haima кровь) — наследственное аутосомно-рецессивное заболевание, обусловленное недостаточностью ферментов, участвующих в обмене галактозы. Основным источником галактозы является лактоза – дисахарид, содержащийся в большом количестве в молоке и молочных продуктах. В кишечнике молочный сахар под действием фермента лактазы расщепляется на глюкозу и галактозу, затем галактоза всасывается и в печени превращается в глюкозу. Впервые заболевание описано в 1908 г. Reuss. Детальное описание галактоземии представил в 1917 г. F. Goppert [3]. Первая подробная клинико-биохимическая картина болезни дана Mason в 1935 г. [5].
Частота галактоземии в разных популяциях колеблется от 1:187000 до 1:18000, частота носителей гена галактоземии (гетерозигот) в популяции составляет 1:300 [1].
В связи с этим представляет интерес клиническое наблюдение – случай ранней диагностики галактоземии у новорожденного в нашей клинике. Девочка Р., родилась у 27-летней женщины, страдающей хроническим пиелонефритом, от второй беременности, которая протекала с угрозой прерывания. Девочка родилась путем кесарева сечения, в удовлетворительном состоянии, с массой тела 2980 г., длиной 50 см, оценка по шкале Апгар на 1-ой и 5-ой мин – 8/9 баллов. С 3-х сут жизни отмечено появление желтухи, которая была расценена как физиологическая, содержание билирубина в сыворотке крови составляло 180– 200 мкмоль/л. Дальнейшее нарастание желтухи до IV–V степеней, увеличение содержания билирубина в сыворотке крови потребовало проведения фототерапии. На 5-е сут жизни был выставлен диагноз: неонатальная желтуха.
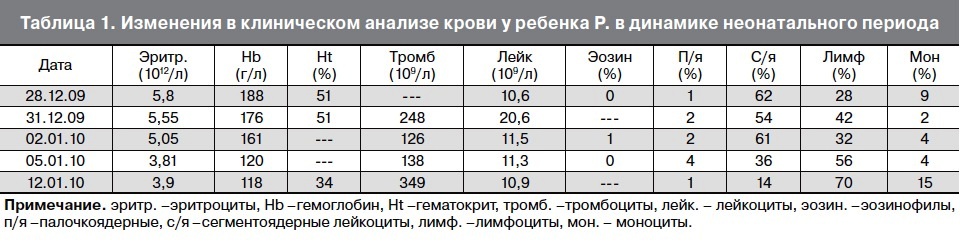
Ребенок находился на грудном вскармливании. В конце раннего неонатального периода состояние ребенка было средней тяжести за счет синдрома угнетения функций центральной нервной системы (гиподинамия, гипотония, гипорефлексия). Потеря массы тела на 7-е сут жизни составила 16% от массы тела при рождении. Желтушность кожных покровов достигала IV–V степеней, уровень билирубина на 7–8-е сут жизни колебался в пределах 257–310 мкмоль/л, в клиническом анализе крови отмечалось нарастание лейкоцитоза от 10,6×109/л, до 20,6×109/л. Динамика клинической картины расценивалась как нарастание инфекционного токсикоза, что явилось показанием к назначению антибактериальной терапии аугментином, продолжена фототерапия.
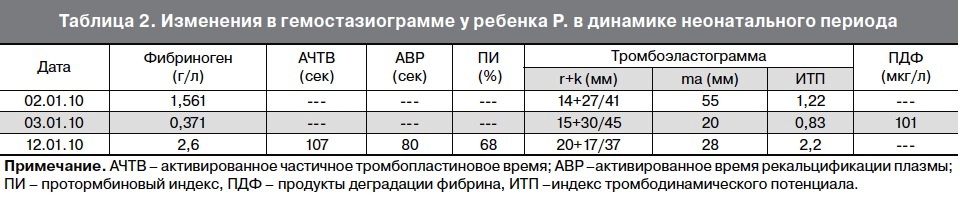
На 9-е сут жизни состояние с отрицательной динамикой за счет нарастания синдрома угнетения функций центральной нервной системы (гипорефлексия, гиподинамия, мышечная гипотония) на фоне сохраняющейся гипербилирубинемии, уровень билирубина в сыворотке крови составил 352 мкмоль/л. Сохранялись признаки инфекционного токсикоза: кожные покровы бледные, с желтушно-сероватым оттенком, выраженные микроциркуляторные нарушения (периоральный цианоз, акроцианоз), отмечались проявления синдрома диссеминированного внутрисосудистого свертывания – ДВС (кровоточивость из мест инъекций). В клиническом анализе крови отмечалась анемия, тромбоцитопения (126×109/л), лейкоцитоз (20,6×109/л), С-реактивный белок – отрицательный.
На 10-е сут жизни состояние ребенка очень тяжелое за счет течения ДВС-синдрома (выраженная гипокоагуляция), гипербилирубинемии, сохранялась гиподинамия, гипорефлексия, сероватый колорит кожных покровов, снижение тургора мягких тканей. При исследовании биохимических показателей крови отмечалась гипербилирубинемия, повышение уровня ферментов печени. Изменение параметров анализов крови в динамике неонатального периода отражено в табл. 1, табл. 2, табл. 3.
Для уточнения локализации инфекционного очага было проведено полное обследование ребенка. Рентгенография органов грудной клетки от 31.12.09 – патологии не выявлено, нейросонография (28.12.09, 11.01.10) – без патологии. Ультразвуковое исследование внутренних органов (02.01, 11.01.10) не выявило патологических изменений. При эхо-кардиографии (02.01.10) – размеры сердца не увеличены, межжелудочковая перегородка интактна, функция клапанного аппарата не нарушена, овальное окно открыто, размер овального окна – 0,2 см. Артериальный проток не функционирует.
Для исключения врожденной цитомегаловирусной (CMV) инфекции проведено исследование антител: CMV IgМ – 18% – результат отрицательный, CMV IgG – 85 МЕ/мл – реакция положительная (материнские IgG).
Ребенок получал антибактериальную терапию, свежезамороженную плазму, иммуноглобулин, антигеморрагическую, антианемическую, гепатопротекторную терапию, фототерапию.
Учитывая нарастание лейкоцитоза, явлений интоксикации, ДВС-синдрома, нельзя исключить течение инфекционного процесса, в связи с чем в терапию был включен иммуноглобулин.
Несмотря на проводимое лечение, состояние ребенка ухудшалось, при этом отсутствовали признаки нарастания инфекционного процесса, было заподозрено врожденное заболевание обмена веществ – галактоземия. Ребенок был переведен на безлактозную смесь нутрилон на 10-е сут жизни.
На 12-е сут жизни в неврологическом статусе сохранялся синдром угнетения центральной нервной системы, отмечалось снижение уровня билирубина (до 213 мкмоль/л), в общем анализе крови воспалительных изменений не было, сохранялась тромбоцитопения. На 16-е сут жизни наблюдалась положительная динамика: уменьшились проявления синдрома угнетения функций центральной нервной системы, сосание стало более активным, весовая кривая стала положительной. На 18-е сут жизни в биохимическом анализе крови отмечена положительная динамика (нормализовались уровни ферментов, общий билирубин снизился до 28 мкмоль/л).
На 19-е сут жизни получены результаты из центра неонатального скрининга, где была выявлена галактоземия – уровень общей галактозы в сыворотке крови 71 мг%.
На 20-е сут жизни ребенок и его родители были обследованы в лаборатории наследственных болезней обмена веществ Медико-генетического научного центра. Выявлено резкое снижение активности галактозо-1-фосфатуридилтрансфе-разы у ребенка – 0,19 Е/rHb (при норме 4,40– 15,00 Е/rHb); у матери – 3,65 Е/rHb, у отца – 3,52 Е/rHb.
Таким образом, учитывая анамнез, клинические и лабораторные данные, был выставлен диагноз: Галактоземия. Анемия. ДВС-синдром.
В результате проводимой терапии (девочку кормили безлактозной смесью нутрилон) состояние ребенка улучшилось: она стала активнее, сосала охотно, назначенный объем питания высасывала, в весе прибавляла. В анализе крови нормализовались уровни тромбоцитов (349×109/л), лейкоцитов (10,9×109/л), проявления анемии сохранялись. В биохимическом анализе крови нормализовались уровни билирубина, печеночных ферментов, щелочной фосфатазы.
Ребенок был выписан домой на 22-е сут жизни в удовлетворительном состоянии. В настоящее время девочке 2,5 года, развитие соответствует возрасту. Соблюдает безлактозную диету, регулярно наблюдается у профильных врачей (генетика, офтальмолога, диетолога, невропатолога), проводится ежегодно ультразвуковое исследование печени, почек и органов брюшной полости (все показатели в норме), биохимический анализ крови (без патологии), анализ на галактозу один раз в полтора месяца, последний показатель 1,6 мг% при полноценном питании. Из препаратов получает элькар, остеокеа.
Таким образом, этот клинический случай демонстрирует раннюю диагностику галактоземии, что позволило достигнуть компенсации врожденного дефекта метаболизма галактозы в возрасте 3 нед и оптимизировать прогноз отдаленного развития ребенка.
Обсуждение клинического случая
В настоящее время описано 3 типа галактоземии, обусловленных соответственно недостаточностью галактозо-1-фосфат-уридилтрансферазы (классический тип галактоземии), галактокиназы и уридилдифосфат-галактозо-4-эпимеразы. Данный клинический случай можно отнести к наиболее распространенному классическому типу галактоземии, при котором значительно снижается активность фермента галактозо-1-фосфат-уридилтрансферазы. При этом вследствие нарушения расщепления галактозы в организме накапливаются галактозо-1-фосфат, галактоза и галакислотитол, оказывающие токсическое действие на центральную нервную систему, печень, почки, кишечник, органы зрения. Следует отметить, что неонатальные формы галактоземии имеют тенденцию к быстрому прогрессированию, тяжелому течению и достаточно быстро приводят к летальному исходу [2, 6].
Данный случай следует отнести к тяжелому течению, симптомы заболевания появились уже в течение первых дней жизни ребенка. Обычно кормление грудным молоком или молочными смесями приводят к рвоте, диарее, нарастают явления токсикоза и эксикоза, быстро развивается гипотрофия. В течение первых дней жизни появляется выраженная и стойкая желтуха, сопровождающаяся увеличением размеров печени, развивается прогрессирующая печеночная недостаточность. Если диагноз не установлен и продолжается кормление молоком, развитие сосудистых коллатералей и асцита наблюдается на 2–3-м мес жизни, в этот же период появляется спленомегалия. [8]. В нашем случае эти патологические признаки не наблюдались, так как отмена молока и введение безлактозной смеси произошло на 10-й день жизни. Нередко, вследствие поражения печени и замедления свертывания крови отмечаются кровоизлияния на коже и слизистых оболочках. При прогрессировании заболевания происходят необратимые изменения центральной нервной системы, рано обнаруживается задержка психомоторного развития, которая с возрастом становится все более выраженной, однако не достигает таких крайних степеней, как при фенилкетонурии. Уже при рождении ребенка может выявиться двусторонняя катаракта, однако чаще она возникает на 4–7-й нед жизни. [6]. Терминальная фаза болезни характеризуется глубоким истощением (кахексией), признаками тяжелой печеночной недостаточности и наслоением вторичных инфекций.
Дифференциальный диагноз мы проводили с гемолитической болезнью новорожденных, обусловленной несовместимостью крови матери и плода по резус-фактору или группам крови системы АВО; с гепатозами, при которых уровень галактозы в крови и моче не повышен; с почечной глюкозурией, при которой уровень глюкозы в крови обычно нормален, печень и селезенка не увеличены; с сепсисом, характеризующимся повышением температуры тела, наличием одного или нескольких очагов инфекции, высевом из крови патогенной флоры; с цитомегалией, при котором в крови выявляется специфический возбудитель.
На основании приказа № 185 от 22.03.2006 Минздравсоцразвития России «О массовом обследовании новорожденных детей на наследственные заболевания», массовый неонатальный скрининг на галактоземию проводят на 4-е сут жизни доношенным новорожденным и на 7-е сут жизни – недоношенным детям. При выявлении уровня общей галактозы 7 мг% и выше проводится повторное исследование. При подозрении на галактоземию применяют скринингтесты: определение содержания галактозы в моче, например с помощью диагностических полосок «PentaPHAN» и «TetraPHAN»(количество галактозы определяют до и после кормления ребенка молоком или молочными смесями, содержащими лактозу). Используется также тест Гатри – полуколичественный метод определения содержания галактозы в крови и моче, основанный на способности определенного штамма кишечной палочки окислять галактозу. Подтверждает диагноз обнаружение низкой активности галактозо-1-фосфат-уридилтрансферазы в эритроцитах и повышенного содержания в них галактозо-1-фосфата. [4]. Возможна пренатальная диагностика болезни путем исследования активности галакислотозо-1-фосфат-уридилтрансферазы в культуре клеток амниотической жидкости, полученной путем амниоцентеза. В сомнительных случаях для диагностики галактоземии может быть использован тест на толерантность к галактозе — определение сахарной кривой после пероральной нагрузки галактозой в количестве 75 г/кг; у больных галактоземией отмечаются высокий подъем и замедленное снижение сахарной кривой. В нашем случае основным методом лечения явилась диетотерапия. Ребенку с подозрением на галактоземию была назначена специализированная безлактозная смесь, своевременно начатая патогенетическая терапия определила прогноз и течение заболевания.



{kind=link}
{kind=link}
{kind=link}