
Эпидермолитический ихтиоз (эритродермия ихтиозиформная буллезная, эпидермолитический гиперкератоз, ихтиоз буллезный, ichthyosis epidermolytica, буллезный тип ихтиозиформной эритродермии, эпидермолитический генерализованный гиперкератоз, буллезный ихтиозиформный гиперкератоз, врожденный универсальный акантокератолиз) – это редкий врожденный кератинопатический ихтиоз, характеризующийся фенотипом, протекающим с образованием пузырей при рождении, который прогрессирует в гиперкератический фенотип. Тип наследования – аутосомно-доминантный, распространенность оценивается в 1:200 000–1:300 000 новорожденных детей, мужчины и женщины болеют одинаково часто [1, 2]. Болезнь обусловлена мутациями генов, кодирующих эпидермальные супрабазальные кератины 1 (KRT1; 12q13.13) и 10 (KRT10; 17q21-q23), которые повреждают промежуточные филаменты кератина в супрабазальных кератиноцитах. Существует определенная связь между генотипом и фенотипом, положение мутации в гене может влиять на тяжесть фенотипа [3]. Буллезные (эпидермолитические; кератинопатические) ихтиозы проявляются такими симптомами, как эритродермия с рождения, эрозии, пузыри на коже, гиперкератоз, имеющий игольчатый вид, повышенная восприимчивость к суперинфекциям, выраженный гиперкератоз (бурого/грязно-серого/буро-черного цвета) кожи в области складок, характерный неприятный запах от больного [4]. Вовлечение ладоней и подошв, как правило, связано с мутациями в гене KRT1.
Гистологическое исследование при данной патологии проявляется межклеточной разобщенностью, дегенеративными изменениями шиповатого слоя эпидермиса, проявляющегося разрушением межклеточных мостиков (участки акантолиза) [5]. При электронной микроскопии выявляются большие зоны перинуклеарного цитоплазматического ретикулума, масса рибосом и митохондрий в клетках зернистого и верхней части шиповатого слоев, утолщение тонофиламентов, вертикальная ориентация эпидермоцитов в роговом слое, дефекты кератинизации [2].
Клинические симптомы заболевания у новорожденного необходимо дифференцировать с токсическим эпидермальным некролизом, наследственным буллезным эпидермолизом, синдромом недержания пигмента, врожденной герпетической инфекцией [1].
В статье представлено клиническое наблюдение ребенка с эпидермолитическим ихтиозом и врожденным пороком развития легкого, родившегося в перинатальном центре.
Клиническое наблюдение
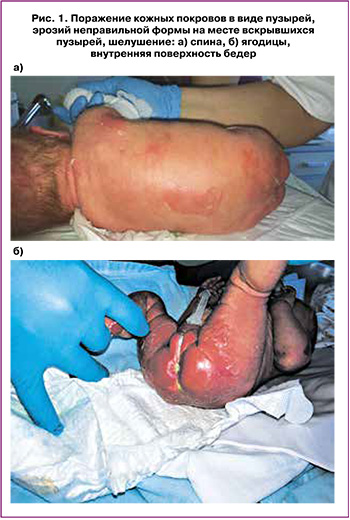 Доношенная девочка у женщины Г. от 3-й беременности, протекавшей на фоне респираторной вирусной инфекции с повышением температуры до 39,4˚С, отеков беременных, протеинурии. Роды 2-е своевременные путем операции кесарева сечения в сроке 38 недель. Родилась доношенная девочка массой тела 2850 г, длиной 51 см. Оценка по шкале Апгар составила 8/8 баллов. При осмотре у ребенка отмечалось поражение кожных покровов в виде пузырей, эрозий неправильной формы на месте вскрывшихся пузырей, шелушения (рис. 1). Проводилась дифференциальная диагностика между инфекционными заболеваниями кожи и наследственной патологией (буллезный эпидермолиз, ихтиоз). Признаков инфекционного токсикоза у ребенка не отмечалось, по результатам микробиологического обследования (посевы из нестерильных локусов, крови, содержимого пузырей, исследование крови методом полимеразной цепной реакции, соскобов из зева и ануса на грам-, грам+ флору) инфекционные поражения кожи были исключены. Антенатально подозрений на врожденный порок легких не было.
Доношенная девочка у женщины Г. от 3-й беременности, протекавшей на фоне респираторной вирусной инфекции с повышением температуры до 39,4˚С, отеков беременных, протеинурии. Роды 2-е своевременные путем операции кесарева сечения в сроке 38 недель. Родилась доношенная девочка массой тела 2850 г, длиной 51 см. Оценка по шкале Апгар составила 8/8 баллов. При осмотре у ребенка отмечалось поражение кожных покровов в виде пузырей, эрозий неправильной формы на месте вскрывшихся пузырей, шелушения (рис. 1). Проводилась дифференциальная диагностика между инфекционными заболеваниями кожи и наследственной патологией (буллезный эпидермолиз, ихтиоз). Признаков инфекционного токсикоза у ребенка не отмечалось, по результатам микробиологического обследования (посевы из нестерильных локусов, крови, содержимого пузырей, исследование крови методом полимеразной цепной реакции, соскобов из зева и ануса на грам-, грам+ флору) инфекционные поражения кожи были исключены. Антенатально подозрений на врожденный порок легких не было.
Ребенок осмотрен дерматологом, на основании клинической картины выставлен диагноз: «Эпидермолитический ихтиоз», который был подтвержден при секвенировании полного экзома – выявлена мутация в гене KRT10.
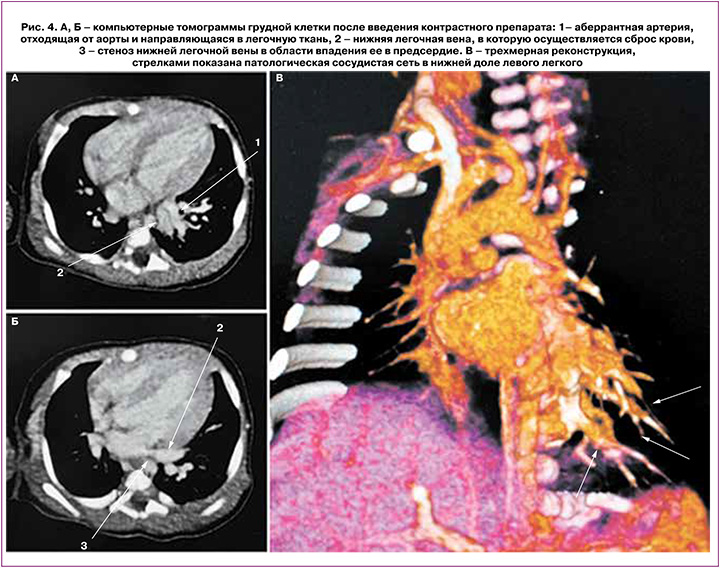
Проводилась местная терапия (асептические повязки на пораженные участки кожи, обработка эмолентами). На фоне проводимого лечения состояние кожи – с положительной динамикой, отмечались очаги гиперкератоза в области стоп, голеней, локтей (рис. 2), кистей. В возрасте 6 суток жизни отмечались воспалительные изменения в общем анализе мочи (лейкоциты покрывают все поля зрения), выставлен диагноз «Инфекция мочевыводящих путей». Проводилась антибактериальная терапия Амписидом в дозе 75–150 мг/кг/сут внутривенно струйно с положительным эффектом, завершена на 12-е сутки жизни. На 14-е сутки жизни состояние ребенка – с отрицательной динамикой за счет появления дыхательных нарушений в виде тахипноэ до 70/мин с втяжением подреберий, тахикардии до 170 /мин, грубого систолического шума с проведением на спину. Учитывая выявленные изменения, проводился контроль эхокардиографии (ЭхоКГ), по результатам которой определялась высокая легочная гипертензия – давление в правом желудочке составляло 85 мм рт. ст., отмечались увеличение и гипертрофия правого желудочка и умеренная гипертрофия левого желудочка сердца. Уровень N-терминального мозгового натрийуретического пропептида (NT-proBNP) составил 38 728 пг/мл. Клинические симптомы дыхательных нарушений сопровождались увеличением количества лейкоцитов в клиническом анализе крови до 19,6×109/л; по данным рентгенографии органов грудной клетки отмечались снижение прозрачности легочных полей с усилением легочного рисунка справа и признаки перегрузки правых отделов сердца. По результатам клинических данных был выставлен диагноз неонатальной пневмонии, в связи с чем проводилась антибактериальная терапия (ванкомицин 30 мг/кг/сут внутривенно капельно, сульперазон 80 мг/кг/сут внутривенно капельно в течение 7 дней). Учитывая клинические признаки сердечной недостаточности, дилятационной кардиомиопатии, сохраняющиеся признаки легочной гипертензии, проводилась диуретическая терапия (верошпирон 3,5–4,0 мг/кг/сут, фуросемид 1–2 мг/кг/сут перорально); на 26-е сутки жизни назначена вазодилатирующая терапия (силденафил-кардио (Ревацио) 1 мг/кг/сут перорально), однако недостаточность кровообращения сохранялась. По данным ЭхоКГ отмечались кардиомегалия, легочная гипертензия – давление в правом желудочке составляло 60 мм рт. ст. На 33-е сутки жизни с диагностической целью выполнена мультиспиральная компьютерная томография органов грудной и брюшной полости с внутривенным контрастированием, выявлен порок развития левого легкого – интралобарный секвестр нижней доли левого легкого с выраженным сбросом крови по аномальному сосуду в венозное русло малого круга кровообращения, расширением нижней легочной вены слева и ее стенозом в области впадения в предсердие; гипоплазия левой легочной артерии; тромбоз нижней полой вены с частичной реканализацией и формированием паравертебральных сплетений (рис. 3, 4).

Таким образом, тяжесть состояния ребенка была обусловлена наличием врожденного порока развития левого легкого с формированием сосудистого шунта из аорты в нижние легочные вены, что явилось причиной легочной гипертензии, перегрузки камер сердца и сердечной недостаточности. Учитывая значительный объем шунтируемой крови, признаки легочной гипертензии, перегрузки камер сердца и сердечной недостаточности ребенок на 39-е сутки жизни был переведен в отделение хирургии новорожденных, где была проведена торакоскопическая перевязка аномального артериального сосуда нижней доли левого легкого. В послеоперационном периоде состояние ребенка – с положительной динамикой, отмечалось уменьшение размеров камер сердца, снижение давления в системе легочной артерии (расчетное давление в правом желудочке по НТК 40–44 мм рт. ст., при системном 118/66 мм рт. ст.). Уровень NT-proBNP в крови снизился до 977,8 пг/мл. На 40-е сутки жизни ребенок переведен в отделение патологии новорожденных и недоношенных детей и на 48-е сутки выписан домой в удовлетворительном состоянии. При ЭхоКГ перед выпиской отмечалось открытое овальное окно, небольшое утолщение миокарда обоих желудочков, без нарастания в динамике, небольшое увеличение левых отделов сердца, умеренная гипоплазия левой легочной артерии, насосная и сократительная функции миокарда сохранены, данных за легочную гипертензию нет.
Клинический диагноз. Основной. Множественные врожденные пороки развития: Эпидермолитический ихтиоз. Врожденный порок развития легкого – интралобарный секвестр нижней доли левого легкого с выраженным сбросом крови по аномальному сосуду.
Сопутствующие. Неонатальная пневмония. Инфекция мочевыводящих путей.
Осложнения. Вторичная легочная гипертензия, ассоциированная форма. Дилятационная кардиомиопатия. Недостаточность кровообращения 1-2А степени.
Проводились регулярная ЭхоКГ и наблюдение у кардиолога. На данный момент ребенку 11,5 месяца, весит 7 кг, рост 72 см. Развивается по возрасту. Со стороны сердца – полости не увеличены, сократительная способность миокарда сохранена, незначительная гипертрофия миокарда правого желудочка.
Обсуждение
В настоящее время известно несколько синдромальных форм, включающих ихтиоз, среди них: сндром Нетертона, включающий наличие атопии, отставание в психическом и физическом развитии [6], синдром Рефсума – наследственно-семейная полиневрическая атаксия, синдром Шегрена–Ларссона (умственная отсталость, нарушение походки, спастическая ди- или тетраплегия, дегенерация сетчатки глаз, дефекты зубов, костей, дерматоглифические аномалии, гигантизм или карликовость, скелетные аномалии, синдром Руда, проявляющийся судорожными припадками и умственной отсталостью [7].
В описанном клиническом наблюдении методом секвенирования экзома выявлена мутация в гене KRT10, однако у родителей ребенка данная мутация не выявлена, что подтверждает ее патогенность. В настоящее время эти исследования являются достаточно трудоемким и дорогостоящим процессом, однако страдающим ихтиозом пациентам и их родственникам необходимо рекомендовать генетическое консультирование для получения более подробной информации о типе наследования, прогнозе и риске развития данной патологии у потомства от родителей пациента и от самого пациента. При обнаружении мутаций, обусловливающих течение тяжелых форм ихтиоза, при планировании беременности рекомендуется обсуждение и выбор дальнейшей тактики. Возможно проведение как предимплантационной, так и пренатальной диагностики с биопсией хориона (12-я или 20-янеделя гестации) или амниоцентезом (начиная с 14-й недели гестации) [8, 9]. Родоразрешение, дальнейшее обследование и ведение новорожденного необходимо проводить в федеральных или региональных перинатальных центрах.
Заключение
Представленное клиническое наблюдение уникально в связи с сочетанием двух врожденных заболеваний у одного ребенка, затрагивающих разные системы организма, и такие ассоциации нуждаются в дальнейшем изучении.


