
Среди всех пороков развития плода врожденные пороки сердца (ВПС) занимают лидирующее место, составляя около 30% в структуре аномалий развития [1]. Частота встречаемости ВПС в популяции составляет от 6 до 8 на 1000 случаев живорождения (0,6–0,8) и 10% от случаев мертворождения [2]. Частота неонатальной смертности от ВПС выше в раннем неонатальном периоде [3]. В Российской Федерации ежегодно рождается около 20 000 детей с ВПС, из них около четверти новорожденных нуждаются в оперативном лечении в первые дни жизни [4].
ВПС классифицируются по степени тяжести, влиянию на легочный кровоток, характеру нарушения гемодинамики. Также выделяют физиологическую и анатомическую классификации [5]. Ряд авторов выделяют изолированные ВПС и ВПС, сочетанные с экстракардиальными аномалиями [6].
В ряде исследований используют классификацию ВПС по Botto L. et al., основанную на фенотипе и этиологии ВПС: 1 – конотрункальные пороки (перерыв дуги аорты, тетрада Фалло, транспозиция магистральных артерий, двойное отхождение сосудов от правого желудочка, общий артериальный ствол), 2 – дефект межпредсердной перегородки, 3 – аномальный дренаж легочных вен, 4 – обструкция выводного тракта левого желудочка (синдром гипоплазии левых отделов сердца (СГЛОС), коарктация/стеноз аорты), 5 – обструкция выводного тракта правого желудочка, 6 – септальные дефекты, 7 – гетеротаксия, 8 – единственный желудочек, 9 – комбинированные дефекты [7].
Этиология ВПС плода разнообразна и недостаточно изучена. По мнению различных авторов, около 20–30% случаев ВПС имеет генетическую причину развития заболевания [8]. Для оценки генетической составляющей, как причины формирования ВПС наиболее целесообразно исследовать группы пороков, имеющие неблагоприятный или сомнительный прогноз с точки зрения детской инвалидизации и смертности.
Доказано наличие ВПС при следующих анеуплоидиях: трисомии хромосомы 18 – 95% [9], хромосомы 13 – 60–80% [10], хромосомы 21 – 40–50% [11], хромосомы 9 – 65–80%, хромосомы 8 – 25%, моносомия X – 25–50% [12], синдром Кляйнфельтера – 50%. В литературе описаны сочетания таких патологических вариантов числа копий генов (CNV) с ВПС, как: 20р12 – 85–94%, 22q11.2 – 75–80% [13], 4р – 50–65%, 7q11.23 – 53–85%, 8р – 50–75%, 5р – 30–60%, 11q – 56%, 10р – 50% [5]. В свою очередь Wang H. et al. в метаанализе описали суммарную долю хромосомных аномалий при ВПС. Так, анеуплоидии, делеции 22q11 и другие CNV у плодов с ВПС составили 23% (95% ДИ 20–26%), 19% (95% ДИ 16–22%), 2% (95% ДИ 2–3%) и 4% (95% ДИ 3–5%) соответственно [14].
Часть ВПС ассоциирована с моногенной патологией. По данным проспективного когортного мета-анализа Mone F. et al. (2021), показано, что при ВПС плода дополнительная диагностическая ценность экзомного секвенирования по сравнению с кариотипированием и хромосомным микроматричным анализом (ХМА) составила около 12,7% [15].
Учитывая вышеизложенное, выявление генетической природы ВПС позволяет определить дальнейшую тактику ведения беременности, сформулировать четкие показания для прерывания беременности, спланировать маршрутизацию беременной.
Цель исследования: изучить частоту встречаемости хромосомных аномалий при различных нозологических формах ВПС плода.
Материалы и методы
На перинатальный консилиум ФГБУ «Национальный медицинский исследовательский центр акушерства, гинекологии и перинатологии имени академика В.И. Кулакова» Минздрава Российской Федерации (ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ) с 2019 г. по 2023 г. обратились 72 пациентки с изолированными ВПС плода, диагностированными по данным ультразвукового исследования (УЗИ) в региональных перинатальных центрах.
В дальнейшем диагноз ВПС плода был верифицирован на основании эхокардиографии сердца плода (ЭХО КГ) специалистами ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ на сроке беременности от 15 до 30 недель. Все супружеские пары были консультированы акушером-гинекологом, кардиохирургом и генетиком в рамках работы перинатального консилиума, где супружеской паре была предоставлена полная информация о диагнозе, прогнозе заболевания. Дизайн исследования представлен на рисунке.
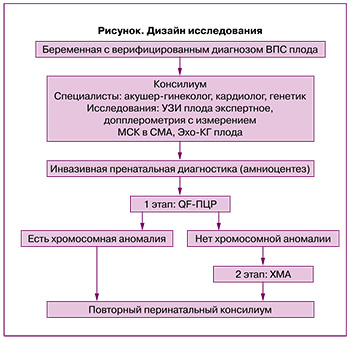
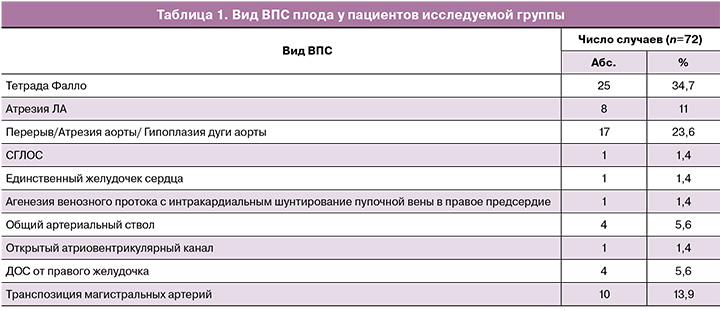
В группу исследования вошли пациентки с такими ВПС плода, как: тетрада Фалло – 25/72 (34,75%), атрезия легочной артерии (ЛА) – 8/72(11%), перерыв дуги аорты, гипоплазия аорты, атрезия аорты – 17/72 (23,6%) случаях, транспозиция магистральных артерий – 10/72 (13,9%), двойное отхождение сосудов (ДОС) от правого желудочка – 4/72 (5,6%), общий артериальный ствол – 4/72 (5,6%), открытый атриовентрикулярный канал – 1/72 (1,4%), СГЛОС – 1/72 (1,4%), единственный желудочек – 1/72 (1,4%), агенезия венозного протока – 1/72 (1,4%) случаев (табл. 1).
Учитывая выявленный ВПС плода, для определения вклада генетического фактора в формирование ВПС плода было предложено проведение инвазивной пренатальной диагностики (ИПД) в сроках от 15 до 30 недель. Обращает на себя внимание тот факт, что у всех беременных были нормальные показатели скрининга I триместра, в связи с чем, данным пациенткам ИПД ранее не проводилась.
Перед вмешательством всеми пациентками было подписано добровольное информированное согласие, которое включало подробную информацию о возможных рисках и осложнениях ИПД. Посредством трансабдоминального амниоцентеза было забрано 40 мл амниотической жидкости. Далее образец направлялся в институт репродуктивной генетики ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ для выполнения хромосомного анализа.
Исследование полученных образцов ДНК плодов проводилось в 2 этапа.
На 1-м этапе проводили выявление аномалий хромосом 13, 18, 21, Х, Y с помощью STR маркеров методом полимеразной цепной реакции (QF-ПЦР) для фрагментного анализа (ТУ 21.20.23-113-46482062-2021) для ускоренного определения наиболее частых анеуплоидий, на долю которых приходится свыше 95% всей хромосомной патологии у новорожденных [16]. Метод QF-ПЦР выполняется в течение 3 суток, в то время как стандартное кариотипирование с выделением ДНК плода из околоплодных вод занимает порядка 3 недель. По данным отечественных авторов, целесообразность выполнения пренатального кариотипа должна обсуждаться индивидуально, так как при нормальном кариотипе не исключено наличие микрохромосомных перестроек, которые в случаях ВПР плода встречаются в 2–3 раза чаще [17]. При выявлении вышеуказанных анеуплоидий, дальнейший диагностический поиск прекращался, при отсутствии патологии хромосом 13, 18, 21, Х, Y материал направлялся на второй этап исследования.
На 2-м этапе исследование ДНК плода проводили с помощью SNP-олигонуклеотидного ХМА. Исследование выполняли с использованием системы GenoScan3000 на микроматрицах CytoScan Optima (Thermo Fisher Scientific, США) согласно протоколу производителя. Анализ полученных данных производился с помощью программного обеспечения ChАS («Сhromosome Аnalysis Suite»). Полученные вариации числа копий (copy number variation, CNV) интерпретировались по пяти категориям, опираясь на критерии ACMG [18]. При интерпретации в соответствии с рекомендациями оценивался размер, место расположения вариаций числа копий генов, генный состав, описание в базах данных (Clinvar, OMIM, ORPHANET, DECIPHER, DGV), а также в литературе. В отчет выносились патогенные и вероятно патогенные вариации числа копий генов, варианты неясной клинической значимости включались только при высоком соответствии с клинической картиной.
Статистический анализ
Статистическая обработка данных выполнялась с помощью таблиц Microsoft Exel (США). Качественные показатели выражены в абсолютных и относительных величинах (%), количественные показатели представлены в виде среднего арифметического (M) и стандартного отклонения (SD).
Результаты и обсуждение
Были проанализированы клинико-анамнестические данные пациентов исследованной группы.
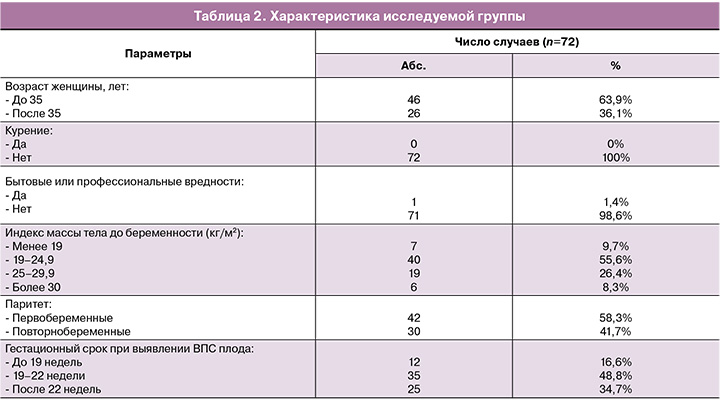
Средний возраст пациентов составил 32,4 лет. Старше 35 лет было 26/72 (36,1%) женщин. Средний срок беременности при выявлении ВПС плода составил 20 недель (SD 3,2, от 15 до 30 недель). В 42/72 (58,3%) случаях пациентки были первобеременными.
При сборе анамнеза не выявлено влияния бытовых и профессиональных вредностей, пациентки не курили и не имели вредных привычек. К бытовым факторам вредности относились: микроклиматические, радиационные, микробиологические и токсикохимические характеристики мест проживания пациенток. Ранняя диагностика ВПС плода до 22 недель имела место в 47/72 (65,2%) случаев, после 22 недель – в 25/72 (34,7%) случаев.
Клинико-анамнестические данные беременных с ВПС плода представлены в таблице 2.
Согласно полученным результатам, ВПС в сочетании с хромосомной патологией выявлен в 16/72 (22,2%) случаев, из них: трисомия хромосомы 21 – 4/72 (5,6%), трисомия хромосомы 18 – 2/72 (2,8%), микроделеция хромосомы 22 – 8/72 (11,1%), микроделеция хромосомы 12 – 1/72 (1,4%), микродупликация хромосомы 1 – 1/72 (1,4%). Анеуплоидии в 6/72 (8,3%) были выявлены методом QF-ПЦР, все вариации числа копий были определены методом ХМА – 10/72 (13,9%) (табл. 3).

В нашей когорте анеуплоидии наблюдались в 8,3% случаев, вариации числа копий генов – в 13,9%, что соотносится с данными американской ассоциации кардиологов (анеуплоидии в 8–10%, вариации числа копий в 3–25% случаев) [19].
Частота синдрома Дауна в нашем исследовании составила 4/72 (5,5%); 3/4 пациенток от дальнейшего пролонгирования беременности отказались, 1 семья приняла решение о пролонгировании беременности, рожденный ребенок погиб в раннем неонатальном периоде в связи с присоединившимся неонатальным сепсисом в сочетании с гипоплазией дуги и перешейка аорты. По данным литературы, около 50% больных синдромом Дауна имеют ВПС [20]. Гены DYRK1A, RCAN1, DSCAM, COLVI, NRIP1, находящиеся на 21-й хромосоме, напрямую участвуют в развитии эндокардиальных подушек [21]. При нарушении слияния эндокардиальных подушек в раннем эмбриональном периоде формируются характерные для синдрома Дауна ВПС: открытый атриовентрикулярный канал, дефект межпредсердной и межжелудочковой перегородки, в некоторых случаях – тетрада Фалло. Важно отметить, что в нашем исследовании вид ВПС плода при синдроме Дауна был аналогичным.
В исследовании в 8/72 (11,1%) случаев ВПС плода хромосомная патология представлена микроделецией 22-й хромосомы; 7/8 пациенток от дальнейшего пролонгирования беременности отказались, 1 пациентка решила пролонгировать беременность, в дальнейшем произошли роды в доношенном сроке. На данный момент ребенок с тетрадой Фалло в хирургическом лечении ВПС не нуждается, проводится дальнейшее обследование и наблюдение.
Критический участок микроделеции 22q11 включает в себя ген TBX1, который играет ключевую роль в формировании 3–4 фарингеальных дуг, из которых развиваются: твердое небо, тимус, верхние отделы сердца (конотрункальная область) и магистральные сосуды [22]. В эмбриогенезе сердца из конотрункальной области развиваются выводной тракт правого желудочка, аорта и легочная артерия. При нарушении развития данной области сердца под действием делеции 22-й хромосомы возникают характерные для синдрома Ди Джоржди ВПС, такие, как: перерыв дуги аорты, тетрада Фалло, транспозиция магистральных артерий, ДОС от правого желудочка, общий артериальный ствол [23]. Вид ВПС плода при микроделеции 22-й хромосомы в нашей когорте пациентов был сопоставим с данными мировой литературы [24, 25].
Делеция 22q11.2 является наиболее частой причиной синдрома Ди Джорджи и ряда других состояний, таких, как велокардиофациальный синдром, синдром конотрункальной аномалии лица, кардиофациальный синдром Кейлера [26]. Синдром Ди Джорджи включает в себя: конотрункальный порок сердца, врожденный иммунодефицит вследствие аплазии или гипоплазии тимуса [27], гипопаратиреоз (причина неонатальных судорог ввиду гипокальциемии), патологию носоглоточного аппарата, нарушение речевого и психического развития.
Иммунодефицит у детей с микроделецией хромосомы 22 связан с гипоплазией или аплазией тимуса, ответственного за выработку Т-клеток, что приводит к частым рецидивирующими вирусными и грибковыми инфекциями. В зависимости от гипоплазии или аплазии тимуса синдром Ди Джорджа классифицируют, как частичный или полный [28].
Также, по данным исследования Brenner M. et al., дети с ВПС в сочетании с делецией 22q11 имеют худшие послеоперационные исходы, обусловленные дефектом гена, кодирующего гликопротеин Ibβ [29]. Известно, что послеоперационные кровотечения возникают в 2 раза чаще, что приводит к повторным хирургическим вмешательствам в первые 24 ч. Данные осложнения связаны с недостатком гликопротеина Ibβ, который необходим для экспрессии комплекса GPIb-V-IX на поверхности тромбоцитов, где он функционирует как рецептор фактора фон Виллебранда. Описанный факт осложняет ведение данной группы пациентов, требующих кардиохирургической помощи, что следует учитывать при прогнозировании выживаемости на этапе пренатального консультирования семьи.
Согласно литературным данным, в 10% случаев делеция 22-й хромосомы наследуется от одного из родителей (наследование происходит аутосомно- доминантным путем); исходя из этого при дальнейшем генетическим консультировании супружеской пары следует предложить прохождение ХМА в формате трио (мать-отец-плод) для выявления наследственных форм патологии сердца для корректного генетического консультирования [30].
В исследовании Mustafa H. et al. (2019) показано, что при ВПС плода хромосомная патология наблюдается в 36,9% случаев, из них в 29,5% – анеуплоидии, в 7,4% – вариации числа копий [31].
В Российской Федерации при проведении ИПД у пациентов с ВПС плода тестом первой линии является стандартное цитогенетическое кариотипирование. Однако авторы Mustafa H. et al. (2019) отмечают, что при использовании ХМА при ВПС диагностическая ценность повышается в 7,4% случаев [31]. Согласно нашему исследованию, в 13,9% случаев ВПС может ассоциироваться с различными микроделеционными синдромами, выявление которых напрямую зависит от метода диагностики. ХМА, как показывает наше исследование, следует считать методом выбора в диагностике генетической патологии у плода с ВПС.
Согласно полученным данным, в 56/72 (77,7%) наблюдений отсутствовала хромосомная аномалия, в 53/72 (73,6%) случаев беременность была пролонгирована, в 1/56(1,9%) случае произошла антенатальная гибель плода на сроке беременности 39 недель, в 1/56 (1,9%) случае беременность прекратила развиваться в 20 недель, в 2/56 (3,6%) случаях произошла гибель новорожденных в раннем послеоперационном периоде.
Проведенное исследование позволило не только определить прогноз для жизни и здоровья плода/новорожденного, но и расширить возможности генетического консультирования данной семьи при последующих беременностях.
Заключение
Проведенное исследование показало, что существует категория пациентов (22%), у которых ВПС сопряжены с хромосомными аномалиями; большая часть из них относится к патогенным вариациям числа копий генов (62,5%). Методом выбора при проведении ИПД является ХМА, в виду того, что ряд микроделеционных/микродупликационных синдромов не выявляется методом стандартного цитогенетического исследования. В нашем исследовании диагностическая ценность ХМА была в 2 раза выше, по сравнению с QF-ПЦР.
Полная информация при проведении пренатальной диагностики необходима для выявления генетических причин ВПС плода.
Своевременное получение информации на пренатальном этапе о сопутствующей хромосомной патологии плода позволяет принять аргументированное решение о пролонгировании или прерывании данной беременности. Дальнейшие исследования позволят расширить возможности генетического консультирования семьи при последующих беременностях и прогнозировать перспективу выживаемости новорожденного.


