
Научно-технический прогресс в биологии и медицине в конце ХХ и начале ХХI вв. привел к появлению новых высокотехнологичных методов ранней диагностики. Это способствовало выявлению причин редких моногенных заболеваний, улучшению профилактики и повышению эффективности лечения многофакторных социально-значимых заболеваний [1].
Создание технологии секвенирования позволило расшифровать последовательность генома человека. С этой целью в 1990 г. Национальным Институтом здоровья США был запущен проект «Геном человека» (The Human Genome Project, HGP), в котором помимо США участвовали Великобритания, Япония, Франция, Германия, Испания и Китай. Проект завершился в 2003 г., когда Национальным центром биотехнологической информации США (NCBI) была опубликована законченная версия сборки генома человека (hg17) [2]. Однако, и данная версия сборки содержала пропуски в последовательности ДНК, которые были впоследствии ликвидированы. Последняя текущая версия генома человека GRCh38.p14 опубликована Консорциумом исследований генома (GRC) 9 мая 2022 г. [3].
Важно отметить, что удельный вклад одного и того же генетического полиморфизма в этиологию и патогенез любого заболевания в разных популяциях и этнических группах может различаться [4, 5]. Поэтому одним из ключевых направлений повышения качества генетической диагностики, основанной на экзомном секвенировании и клиническом генетическом паспорте, является развитие, прежде всего, популяционных баз данных аллельных частот генетических вариантов (в том числе отечественных), играющих роль в патогенезе наследственных и мультифакториальных заболеваний (МФЗ), совершенствование биоинформатических и статистических протоколов обработки и анализа данных секвенирования, особенно в плохо изученных популяциях [6].
Преимущества и недостатки различных технологий NGS
Технологию NGS (Next Generation Sequencing – секвенирование нового поколения) можно разделить на секвенирование c короткими (от 100 до 600 пар нуклеотидов) и длинными (до 900 КБ) прочтениями. В настоящее время наиболее часто применяются методы с короткими прочтениями последовательностей, поскольку они дешевле и имеют более высокую точность, чем методы с длинными прочтениями. Однако это не всегда так. В частности, нами было показано, что при большом покрытии (около 4000Х) применимость данных секвенирования нанопор для определения вариантов митохондриального генома полностью согласуется с данными, полученными на секвенаторах фирмы Illumina [7]. При этом технологии с короткими прочтениями не могут использоваться для секвенирования повторяющихся или гетерозиготных последовательностей, для которых применяются технологии секвенирования с длинными прочтениями [8]. Особенности различных применяемых в медицинской практике технологий NGS представлены в таблице.
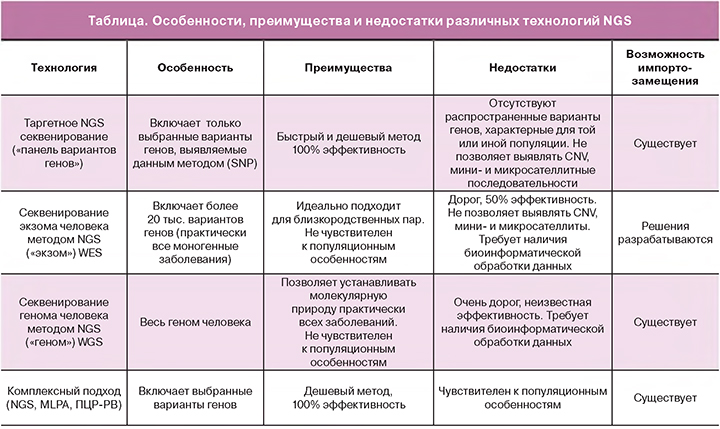
Таргетное секвенирование стало одним из первых практических приложений NGS. Благодаря этой технологии стало возможным прочитывать отдельные гены в 1000 раз дешевле, чем ранее, с использованием полимеразной цепной реакции для длинных фрагментов, позволяющей амплифицировать участки до 50–100000 пар оснований. Данный подход особенно эффективен для анализа небольших генов [9, 10]. Полногеномное секвенирование не так распространено в практике, хотя позволяет провести полный анализ всех генов человека. По данным опубликованной версии генома человека GRCh38, структурные гены, кодирующие белки (экзом), составляют всего 3,09% (90 млн пар нуклеотидов) от общего количества генов, однако содержат около 85% вариантов, ассоциированных с фенотипическим проявлением заболевания [11, 12]. Несмотря на кажущееся преимущество полногеномного подхода над экзомным, анализ диагностической ценности двух этих методов для поиска клинически значимых мутаций не показал такой закономерности. Полногеномное тестирование позволяет выявлять, по разным оценкам, от 44 до 50% всех искомых мутаций, тогда как экзомное уступает ему всего на 2%. Во многом это связано с тем, что при анализе экзома используют более длинные фрагменты и более совершенные конструкции зондов [13]. Поэтому в настоящее время в совокупности с относительно низкой ценой экзомное секвенирование выглядит более привлекательным для клинического применения, так как позволяет обнаруживать редкие патологические генетические варианты (SNP, инсерции, делеции), которые могут служить причиной заболевания [14].
Области и особенности клинического применения NGS
Благодаря экзомному секвенированию достигнут существенный прогресс в диагностике орфанных (редких) заболеваний, более чем в 10 раз возросла эффективность диагностики патогенных вариантов некоторых олигогенных заболеваний, например, MODY-диабета [9]. С помощью новых технологий стало возможным уточнить частоты распространенности многих моногенных заболеваний. Возможно, это связано с тем, что заболевания органов зрения часто диагностируются в пожилом возрасте. В эпоху до NGS болезнь Штаргардта практически не изучали, а клиницисты проблему падения зрения связывали с возрастными изменениями.
Редкие заболевания поражают примерно 1 из 20 человек, но лишь небольшое количество из этих пациентов получают генетический диагноз [15]. Известно примерно 10 000 редких заболеваний, но лишь для менее половины из них известна генетическая причина [16].
В пилотном исследовании проекта «100 000 английских геномов» были получены данные секвенирования полного генома (WGS) для 13037 участников, из которых у 9802 было диагностировано редкое заболевание [17]. В данном исследовании продемонстрировано, что некоторые наши знания о природе наследования: доминантное или рецессивное (например, для гена GP1BB) не всегда могут быть верными [17]. Поэтому результаты подобного рода исследований необходимо учитывать в дальнейшей работе не только ученых (обновление баз данных, информации и т.д.), но и врачей-генетиков.
Важно отметить, что несмотря на то, что секвенирование по Сэнгеру менее производительно и не так экономически выгодно, как NGS, оно продолжает иметь актуальность для ряда практических задач и чаще всего используется для поиска новых или известных мутаций в сравнительно небольшой области молекулы ДНК (от 100 до 1200 пар оснований) и в качестве «золотого стандарта» для подтверждения мутаций, найденных у пациента методом NGS. С помощью секвенирования по Сэнгеру удобно осуществлять поиск мутаций в «горячих точках» хорошо изученных генов, определять нуклеотидную последовательность коротких (содержащих мало экзонов) и безинтронных генов, изучать короткие повторы [1], а также при поиске «второго» патогенного варианта после или вместе с NGS [18].
Основными ограничениями применения технологий целевого секвенирования в клинической практике является их высокая стоимость и сложность в клинической интерпретации данных секвенирования, особенно для объяснения этиологии, патогенеза и симптомов многофакторных заболеваний, на развитие которых кроме генетических факторов сильно влияют внешние факторы и образ жизни [14, 19]. Как и любая другая технология, экзомное секвенирование имеет ряд недостатков, связанных именно с техническими особенностями. Так, получение большого объема данных при WGS и WES связано с проблемой их анализа и обработки: часто бывает трудно отличить небольшие генетические варианты от случайных ошибок, возникающих в процессе секвенирования [20]. Кроме того, основным ограничением WES является неравномерность покрытия зондами последовательностей генов-мишеней генома, что приводит к появлению областей с низким охватом, которые в результате последующего анализа препятствуют точному аннотированию, или пропуску вариантов [21].
Изменение генетической терминологии
Необходимо отметить существенно важное изменение в генетической терминологии, связанное с использованием новых методов. Вместо широко распространенных терминов «мутация» и «полиморфизм» в 2015 г. в США Американским колледжем медицинской генетики и геномики (American College of Medical Genetics and Genomics – ACMG), а в 2017 г. – Российским обществом медицинской генетики было рекомендовано использовать термин «вариант нуклеотидной последовательности» со следующими пятью характеристиками: патогенный (pathogenic); вероятно патогенный (likely pathogenic); неопределенного значения (uncertain significance); вероятно доброкачественный (likely benign); доброкачественный (benign) [22–24]. Данную терминологию активно стали применять не только в диагностическом плане [24], но и при проведении различных исследований.
Сегодня есть четкое понимание, что генетический вариант является основным носителем предикции патогенности заболеваний с двумя основными характеристиками – пенетрантностью (процент носителей соответствующего генотипа, у которых проявляется признак; экспрессивностью (варьирующее проявление признака у особей с одинаковым генотипом.
Таким образом, термины, предложенные еще в 1925 г. Тимофеевым–Ресовским, оказались настолько важными и опережающими свое время [25], что сейчас их сущность позволяет объяснить, почему такой термин, как мутация, уходит в прошлое, и теперь остается только термин «вариант» в пяти состояниях [24].
Биоинформатика – ключевой инструмент в NGS
Биоинформатическая обработка является также частью технологии NGS. Поэтому установление генетической природы заболевания во многом зависит от качественного биоинформатического протокола анализа данных секвенирования [13]. Необходимо учитывать, что в референсной последовательности встречаются ошибки, связанные с так называемыми референсными минорными вариантами – RMA (позициями референсного генома, в которые инкорпорирован редкий или даже патогенный вариант). Такие ошибки необходимо корректировать при проведении биоинформатического анализа [26, 27].
Разработанные биоинформатические алгоритмы вместе с новыми подходами по интерпретации данных позволяют нам описывать не только известные варианты, но и новые [28–30].
NGS и предиктивная медицина
Основными звеньями работы с пациентом должны стать предикция (предсказание) риска заболевания, доклиническая диагностика с определением его стадии и как можно ранее адекватное вмешательство (фармакология, питание и др.) с целью превенции развития заболевания или его перехода в тяжелую стадию. Эти принципы легли в основу стратегии «трех П» – предиктивной, превентивной и персонализированной медицины [31]. Следует отметить, что становление терминов «персонализированной медицины» и «генетического паспорта» появились в Санкт-Петербурге еще в 2000 г., и постепенно происходит их внедрение в общественное сознание [1].
Важно отметить, что большинство болезней не являются моногенными, поэтому прежде чем оценивать риск заболевания, нужно установить его природу: полигенное, олигогенное или МФЗ? Иногда ответить на этот вопрос достаточно сложно. Исходя из нашего опыта [10, 29, 30] и данных литературы, олигогенными можно назвать практически все наследственные болезни [32, 33]. Все определяется вкладом определенных вариантов генов, их экспрессивностью и пенетрантностью. Еще одной сложностью и важной особенностью результатов современных NGS-методов исследования является то, что мы можем выявлять сразу несколько наследственных заболеваний у одного человека. Такие находки создают трудности для клинической интерпретации, но крайне важны при ведении пациентов [34].
Анализ всего экзома может служить разумным подходом для выявления генетических маркеров, в том числе и сложного заболевания и даже в ограниченных выборках. Используя стратегию многоаспектного анализа, мы обнаружили несколько локусов-кандидатов и SNP, которые могут играть важную роль в патогенезе сахарного диабета 2 типа и ожирения у населения России [27]. Таким образом, результат нашего исследования показал, что рациональная фильтрация и ранжирование потенциально «причинных» вариантов может помочь в идентификации генов МФЗ и демонстрирует эффективность технологий секвенирования экзома для поиска новых маркеров МФЗ в когортах ограниченного размера в плохо изученных популяциях [26].
Геномная медицина может также помочь выявить редкие состояния, которые скрыты в рамках сложной диагностики заболевания. Более того, для различных распространенных заболеваний были идентифицированы гены, в которых редкие варианты у гетерозиготных носителей повышают риск в несколько раз. Важным примером является наличие вариантов риска семейной гиперхолестеринемии у 0,4% населения, что повышает риск развития ишемической болезни сердца в 3 раза [35]. Поэтому именно использование экзомного секвенирования может позволить ответить на вопросы оценки риска не только моногенных, но и МФЗ.
Недавнее исследование, проведенное Khera et al., продемонстрировало, как геномные оценки риска можно успешно использовать для характеристики индивидуальной предрасположенности к пяти общим заболеваниям, показав, что 20% населения имели трехкратное увеличение полигенного риска для одного или нескольких заболеваний. Этот уровень риска был сопоставим с тем, который обеспечивали редкие варианты с высокой патогенностью, связанные с моногенными расстройствами [36].
Заключение
Разработка научных основ точной медицины, как для изучения, диагностики и лечения моногенных болезней, так и олигогенных, мультифакториальных и инфекционных заболеваний будет определяться эффективностью использования NGS технологий с учетом современных алгоритмов анализа и классических генетических понятий экспрессивности и пенетрантности.


