
В последние годы в нефрологии неуклонно возрастает интерес к изучению преэклампсии (ПЭ), которую можно считать самой частой причиной гломерулярного поражения почек [1, 2]. Ежегодно в мире регистрируется более 8 млн случаев ПЭ, которая является основной причиной материнской и перинатальной смертности, унося жизни 60 тысяч молодых женщин ежегодно [3].
В настоящее время широко обсуждается роль дисбаланса проангиогенных и антиангиогенных факторов в качестве важного механизма развития артериальной гипертензии (АГ) и протеинурии у пациенток с ПЭ. Установлено, что в момент ПЭ наблюдается дефицит васкуло-эндотелиального фактора роста (VEGF), вызванный циркулирующими рецепторами к VEGF, идентифицированными как растворимые fms-подобные тирозинкиназы-1 – sFlt-1, которые ишемизированная плацента начинает синтезировать за 5–6 недель до клинической манифестации ПЭ. Эти факторы ингибируют как VEGF, так и плацентарный фактор роста – PlGF, обеспечивающие нормальное развитие и функцию плаценты, и, циркулируя в кровотоке матери, могут вносить свой вклад в развитие системной эндотелиальной дисфункции, лежащей в основе всех клинических проявлений ПЭ [4, 5].
Нефрологические проявления ПЭ
Основными нефрологическими проявлениями ПЭ являются АГ, ПУ и/или нарушение функции почек. Поскольку отеки возникают у 60% женщин с физиологически протекающей беременностью, сами по себе отеки перестали рассматриваться как признак ПЭ.
В 2000 году рабочая группа национальной программы изучения АГ (NHBPER) подтвердила, что критериями АГ беременных считается повышение АД свыше 140/90 мм рт. ст., измеренное два и более раз в течение 4–6 часов, особенно у женщин после 20 нед. гестации с предшествующим нормальным АД. Было также высказано мнение, что увеличение систолического АД на 30, а диастолического АД на 15 мм рт. ст. от исходного даже в случае АД менее 140/90 мм рт. ст. обязывает наблюдать женщину для исключения развития ПЭ.
ПУ может предшествовать гипертензии, но обычно развивается одновременно или следом за ней. Важнейшим диагностическим критерием ПЭ считается уровень ПУ от 0,3 г/сут и более. ПУ отличается быстрым (иногда почасовым) нарастанием. Вследствие этого гипопротеинемия (у беременных альбумин в крови менее 25 г/л) и нефротические отеки могут отсутствовать в первые дни развития ПЭ [6]. ПЭ считается ведущей причиной развития нефротического синдрома (НС) во время беременности [7], хотя этот факт практически не известен нефрологам. Однако он был подтвержден еще в 1977 г. К.А. Fisher и соавт., выполнявшими биопсию почки беременным женщинам с ПЭ в связи с развитием НС и в 67% случаев обнаружившими в биоптатах лишь ее гистологические признаки [8]. В целом НС является довольно редким осложнением беременности, частота которого не превышает 0,0025% [9], однако до настоящего времени отсутствуют исследования, сравнивающие частоту НС в зависимости от сроков развития ПЭ.
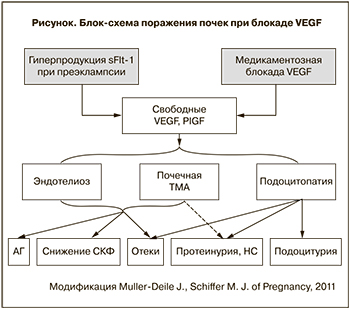 В последние годы обсуждается роль повреждения подоцитов – эпителиальных клеток почечных клубочков, ответственных за гломерулярную проницаемость – в генезе массивной ПУ, в том числе и при развитии ПЭ [10, 11]. При изучении подоцинурии как маркера подоцитопатии отмечена высокая специфичность и чувствительность показателя. По данным V.D. Garovic и соавт. подоцинурия выявлена у 15 из 15 женщин с ПЭ, отсутствовала – у 16 из 16 женщин с неосложненной беременностью, а также – у 7 женщин с другими причинами АГ, ПУ или почечными заболеваниями. Помимо подоцина показателями подоцитопатии могут служить и другие подоцитарные белки: нефрин, подокаликсин, синаптоподин. Однако именно подоцин обладает максимальной специфичностью и чувствительностью и справедливо претендует на место раннего предиктора ПЭ [12].
В последние годы обсуждается роль повреждения подоцитов – эпителиальных клеток почечных клубочков, ответственных за гломерулярную проницаемость – в генезе массивной ПУ, в том числе и при развитии ПЭ [10, 11]. При изучении подоцинурии как маркера подоцитопатии отмечена высокая специфичность и чувствительность показателя. По данным V.D. Garovic и соавт. подоцинурия выявлена у 15 из 15 женщин с ПЭ, отсутствовала – у 16 из 16 женщин с неосложненной беременностью, а также – у 7 женщин с другими причинами АГ, ПУ или почечными заболеваниями. Помимо подоцина показателями подоцитопатии могут служить и другие подоцитарные белки: нефрин, подокаликсин, синаптоподин. Однако именно подоцин обладает максимальной специфичностью и чувствительностью и справедливо претендует на место раннего предиктора ПЭ [12].
При физиологической беременности скорость клубочковой фильтрации (СКФ) увеличивается на 40–60% в течение первого триместра, достигая 140–170 мл/мин [13], в результате чего в сыворотке крови снижается концентрация креатинина, мочевой кислоты, мочевины. При развитии ПЭ СКФ снижается на 30–40% по сравнению с нормальной беременностью, однако уровень сывороточного креатинина практически всегда соответствует референсным значениям небеременных женщин и редко превышает 90–100 мкмоль/л [14], что, в свою очередь, приводит к недооценке почечного повреждения. В связи с этим «золотым стандартом» определения функции почек во время беременности считается определение СКФ методом Реберга–Тареева. Расчетные формулы не приемлемы для применения у беременных, так как, например, СКФ, рассчитанная по формуле MDRD, существенно занижает значения, а формула Кокрофта—Голта, напротив, завышает их [15, 16].
Другим характерным признаком снижения почечной функции при ПЭ является прогрессирующее повышение сывороточного уровня мочевой кислоты – более 500 мкмоль/л. Гиперурикемия часто предшествует протеинурии и обусловлена ухудшением почечной перфузии [17].
Роль VEGF в развитии почечной патологии
VEGF – сигнальный белок, вырабатываемый клетками для стимулирования васкулогенеза (образование эмбриональной сосудистой системы) и ангиогенеза (рост новых сосудов в уже существующей сосудистой системе). Наиболее важную роль в организме человека играет белок семейства VEGF, называемый VEGF-A. В данное семейство также входят плацентарный фактор роста – PlGF и белки VEGF-B (эмбриональный ангиогенез тканей миокарда), VEGF-C (ангиогенез лимфатических сосудов), VEGF-D (развитие лимфатических сосудов в легких). Все члены семейства белков VEGF функционируют, связываясь с двумя близкими по строению мембранными тирозинкиназными рецепторами: рецептором-1 VEGF (VEGFR1 или Flt-1) и рецептором-2 VEGF (VEGFR2 или Flk-1) и активируя их. Эти рецепторы экспрессируются эндотелиальными клетками [18].
Белок VEGF-A связывается с рецепторами VEGFR-1 и VEGFR-2, при этом рецептор VEGFR-2 выступает как посредник почти во всех известных реакциях клетки на VEGF. VEGFR−1 также может выступать как «пустой» рецептор, изолируя белок VEGF от рецептора VEGFR-2 (что представляется особенно важным при васкулогенезе в зародыше).
Экспрессия VEGF стимулируется множеством проангиогенных факторов, включая эпидермальный ростовой фактор, основной фактор роста фибробластов, тромбоцитарный фактор роста и интерлейкин-1. Кроме того, уровни VEGF непосредственно регулируются такими факторами окружающей среды, как рН, давление и концентрации кислорода. Общее влияние этих различных факторов заключается в опосредованной через VEGF стимуляции важных для ангиогенеза факторов, включая антиапоптотические белки, молекулы клеточной адгезии и металлопротеиназы. Однако основным стимулом экспрессии и/или продукции VEGF является именно гипоксия [19].
В физиологических условиях основными функциями VEGF-А являются:
- стимуляция пролиферации эндотелиальных клеток и их дифференциация;
- увеличение сосудистой проницаемости;
- опосредованная эндотелий-зависимая вазодилатация;
- поддержание жизнеспособности эндотелия путем предотвращения апоптоза эндотелиальных клеток;
- участие в ремоделировании экстрацеллюлярного матрикса путем индукции экспрессии активатора плазминогена и PAI;
- усиление экспрессии молекул адгезии на поверхности эндотелиальных клеток [20].
VEGF экспрессируется в почке подоцитами и играет также важнейшую локально-почечную роль, а именно: регуляцию клубочковой проницаемости, образование и поддержание фенестрации эндотелия капилляров клубочков и поддержание цитоскелета подоцитов. Сегодня получены убедительные доказательства того, что при ПЭ дефицит гломерулярного VEGF играет ключевую роль не только в генезе ренальной дисфункции, но и ПУ и АГ. Как оказалось, подоцитарный VEGF обладает не только паракринной функцией в отношении эндотелиальных клеток, но и аутокринной – в отношении самих подоцитов [21]. В связи с этим есть основания полагать, что дефицит VEGF приводит к повреждению подоцитов, распластыванию их ножек, следствием чего и является ПУ [21, 22].
Патогенез почечной патологии при ПЭ
В начале XXI века в ряде эпидемиологических исследований было убедительно продемонстрировано, что женщины, перенесшие ПЭ во время беременности, в последующем имеют высокий риск развития АГ, ишемической болезни сердца, инсульта, в связи с чем ПЭ была отнесена к факторам риска сердечно-сосудистых заболеваний [23].
Принимая во внимание сходство факторов риска сердечно-сосудистых осложнений и хронической болезни почек (ХБП), по-видимому, ПЭ можно рассматривать также и как фактор риска ХБП. Между тем механизмы хронизации почечного поражения при ПЭ, особенно ее раннем развитии – до 34 недели беременности, практически неизвестны нефрологам.
В 2008 г. учеными из Норвегии было опубликовано крупное исследование, показавшее, что рождение маловесных детей сопряжено с риском развития в последующие годы ХБП у матерей [24]. Эта уникальная работа, проводившаяся в течение почти 40 лет, позволила проанализировать связь частоты развития терминальной почечной недостаточности и перенесенной ПЭ. К сожалению, в ней не учитывались сроки развития ПЭ. Оказалось, что риск развития хронической почечной недостаточности у женщин, перенесших ПЭ, был в 4 раза выше, чем в популяции. У 20–40% женщин, не имевших болезней почек до беременности, в течение нескольких лет после родоразрешения сохранялись микроальбуминурия и повышенные цифры АД. Факт того, что у 20–40% пациенток после ПЭ персистирует микроальбуминурия, указывает на возможность необратимого гломерулярного повреждения [2]. Кроме того, ПУ, в том числе и микроальбуминурия, сама по себе вызывает прогрессирующую почечную дисфункцию за счет усиления интерстициального воспаления. [25]. По литературным данным, до 20% женщин, перенесших ПЭ, имеют признаки ХБП после родоразрешения [26, 27].
Конечно, правомерен вопрос, не является ли формирование нефропатии в послеродовом периоде следствием не ПЭ, а почечного заболевания, не диагностированного до наступления беременности. И в ряде случаев получить однозначный ответ не представляется возможным.
Безусловно, достижением последних лет стало предположение о важной роли дисбаланса плацентарных проангиогенных и антиангиогенных факторов как одного из основных механизмов развития ПЭ.
Предпосылками для изучения факторов ангиогенеза при ПЭ явилось сходство клинических проявлений последней и побочных эффектов анти-VEGF-терапии при лечении злокачественных опухолей, которая, как оказалось, может индуцировать развитие ренальной тромботической микроангиопатии (ТМА) [28, 29]. В 2002 г. впервые были опубликованы данные клинических испытаний бевацизумаба – ингибитора VEGF, применение которого приводило к гипертензии и ПУ у больных со злокачественными опухолями разных локализаций [30]. В 2008 г. V. Eremina и соавт. представили шесть клинических наблюдений пациентов со злокачественными новообразованиями, получавших терапию бевацизумабом. Авторы оценивали функцию почек, уровень суточной ПУ до лечения и вскоре после назначения препарата. У всех констатировано ухудшение функции почек с нарастанием ПУ, возникновением АГ в ближайшие месяцы от начала терапии, что явилось показанием к выполнению биопсии почки. Во всех шести нефробиоптатах выявлена картина ТМА, особенностью морфологической картины было сочетание ТМА с распластыванием малых отростков подоцитов, более выраженным у больных с массивной ПУ. После отмены бевацизумаба функция почек нормализовалась у всех пациентов [28]. Авторы высказали предположение, что снижение уровня VEGF в почке в результате блокады его антителами могло привести к локальной почечной дисфункции эндотелия (у всех больных имелись лишь почечные проявления ТМА и отсутствовали ее системные проявления) вследствие нарушения взаимодействия VEGF со своими рецепторами, экспрессируемыми эндотелиальными клетками клубочков. Сходство клинических проявлений побочных эффектов анти-VEGF терапии и ПЭ привело к тому, что некоторые авторы называют ренальные последствия терапии ингибиторами ангиогенеза «преэклампсия-подобным синдромом» [31].
В 2003 г. S.E. Maynard и соавт. установили наличие дефицита VEGF у пациенток в момент ПЭ. Однако в этом случае ингибиция VEGF была вызвана растворимыми рецепторами к VEGF – VEGFR1, идентифицированными как так называемая растворимая fms-подобная тирозинкиназа-1 (sFlt-1), которую синтезирует ишемизированная плацента. Установлено, что избыточный синтез sFlt-1 начинается за 5–6 недель до клинической манифестации ПЭ. Этот фактор ингибирует как VEGF, так и PlGF, обеспечивающий васкулогенез, и, циркулируя в кровотоке матери, может вносить свой вклад в развитие системной эндотелиальной дисфункции, лежащей в основе всех клинических проявлений ПЭ. При этом авторы считают дискутабельным вопрос о ведущем значении нарушения плацентации в патогенезе ПЭ, так как избыток sFlt-1 является самостоятельным фактором развития всех клинических проявлений ПЭ, лежащих в основе так называемого «материнского синдрома» [32].
В почках поражение эндотелия представлено картиной гломерулярного капиллярного эндотелиоза с отеком эндотелиальных клеток, утратой ими фенестр и отслойкой от базальной мембраны, приводящими к окклюзии просвета капилляров, что позволяет рассматривать поражение почек при ПЭ как особый тип ТМА, несмотря на редкость тромбозов капиллярных петель клубочков [7, 29].
У больных с ранними и тяжелыми формами ПЭ при биопсии почки в послеродовом периоде в 35–71% случаев выявляется фокально-сегментарный гломерулярный склероз (ФСГС), носящий, как правило, вторичный характер [33]. Среди механизмов развития ФСГС при ПЭ предполагают роль гломерулярного эндотелиоза, внутриклубочковой гипертензии и гиперкоагуляции. У 20–30% пациенток с выявленным после перенесенной ПЭ ФСГС персистирует АГ, тогда как ПУ отсутствует или выражена минимально. У этих женщин при нефробиопсии в динамике гистологические проявления ФСГС сохраняются даже при отсутствии прогрессирования клинических признаков, хотя явления эндотелиоза постепенно исчезают. В отличие от двуконтурности базальных мембран клубочка (БМК) при мембрано-пролиферативном гломерулонефрите 1-го типа, образование которой происходит за счет интерпозиции мезангиальных клеток между эндотелием и базальной мембраной, при ТМА двуконтурность БМК обусловлена ее расщеплением за счет отложения фибрина в субэндотелиальном пространстве. Течение ФСГС у женщин, перенесших ПЭ, более благоприятное, чем течение первичного ФСГС. Другой морфологической находкой, обнаруживаемой при тяжелом течении нефропатии беременных, является фибриноидный некроз и склероз междольковых артерий почек. Эти изменения являются результатом прямого повреждающего действия фульминантного развития тяжелой или злокачественной гипертензии в момент ПЭ. В отдаленном послеродовом периоде у 75% женщин со склерозом междольковых артерий сохраняется устойчивая АГ, нередко с признаками озлокачествления. Очевидно, что именно ФСГГ и склероз внутрипочечных артерий лежат в основе «остаточных изменений» после перенесенной нефропатии беременных, протекающих обычно под маской «гипертонической болезни» или «гипертонического нефрита».
В последние годы получены данные о том, что умеренно выраженный эндотелиоз, считающийся ранее патогномоничным признаком ПЭ, выявляется у 1/3 пациенток с гестационной АГ даже в отсутствие признаков ПЭ. Более того, минимальные гистологические признаки эндотелиоза в виде очагового отека эндотелиальных клеток встречаются у здоровых беременных, что позволило предположить, что патология эндотелиальных клеток клубочка, выраженная минимально, может быть характерна для беременности вообще [29, 34].
В 2007 г. С.A. Karumanchi и соавт. опубликовали данные, полученные на экспериментальных моделях беременных и небеременных крыс, у которых вызывали ПЭ путем введения в вену sFlt-1. Повышенная концентрация в кровотоке sFlt-1 как у беременных, так и у небеременных животных вызывала АГ и ПУ, что напоминало «человеческую» ПЭ. Гистологические исследования ткани почек этих животных указывали на наличие клубочкового эндотелиоза, характерного для ПЭ. При введении рекомбинантного VEGF121 крысам с клинической картиной ПЭ проявления последней быстро регрессировали, демонстрируя дозозависимый эффект [35].
Эти данные подтверждают результаты совместных исследований корейских и американских ученых, продемонстрировавших в 2001 г. регресс индуцированной почечной ТМА у крыс после введения рекомбинантного VEGF121. Свои данные ученые подтверждали в том числе морфологически [36]. Таким образом, блокада VEGF является одним из важнейших патогенетических механизмов гломерулярного повреждения, лежащего в основе «нефрологической» составляющей ПЭ [28, 37].
Исследование E. Robinson и соавт. [38] продемонстрировало, что ингибиция VEGF седиранибом – сильнодействующим ингибитором VEGFR-2 – в течение трех дней индуцирует у пациентов повышение АД. В качестве объяснения этого феномена авторы предложили блокаду VEGF. Точный механизм АГ при дефиците VEGF не известен, однако B. Li с соавт. удалось показать, что VEGFR-2 является основным посредником для осуществления гипотензивного эффекта VEGF (вазодилатация через механизм высвобождения эндотелиальными клетками оксида азота, простациклина) [32] и ингибирование VEGF способно привести к АГ. Обсуждается также уменьшение площади микроциркуляторного русла вследствие недостатка VEGF как возможная причина АГ. Уменьшение микрососудистого кровотока приводит к увеличению периферического сосудистого сопротивления и снижению концентрации оксида азота. Независимо от этого, VEGF оказывает гипотензивный эффект, воздействуя на барорецепторы эндотелиоцитов [39].
Кроме того, блокада VEGF способна вызвать его дисбаланс с эндотелином, который является мощным вазоконстриктором [40].
Поскольку VEGF необходим для поддержания функционирования подоцитов и регуляции клубочковой фильтрации, его дефицит способен вызвать подоцитопатию и, как результат, ПУ, снижение фильтрационной функции почек, а также гломерулярный эндотелиоз [7] – патогномоничный морфологический признак ПЭ. Установлено, что инактивация одного аллеля гена VEGF ведет к развитию выраженного гломерулосклероза с повреждением подоцитов и эндотелиальных клеток и, как следствие, терминальной почечной недостаточности эмбриона крыс примерно на 9-й неделе гестации [41].
Немногочисленные пока исследования продемонстрировали снижение экспрессии нефрина и синаптоподина в клубочках почек женщин, умерших от ПЭ, и у экспериментальных животных при введении им sFlt-1 или анти-VEGF-антител [42], а также наличие подоцитов в моче женщин с ПЭ [42, 43], что подтверждает предположение о роли подоцитарного повреждения в генезе преэклампсической ПУ. Таким образом, на сегодняшний день существует немало доказательств того, что ингибиция VEGF рецепторами sFlt-1 или анти-VEGF-препаратами способна вызвать гломерулярное поражение. Применительно к ПЭ можно предполагать, что это поражение сочетает в себе гломерулярный эндотелиоз и подоцитопатию (рисунок).
Заключение
Сегодня достигнуты серьезные успехи в изучении патогенеза ПЭ, в том числе механизмов почечного повреждения: доказано, что ишемизированная плацента синтезирует в избытке sFlt-1 – растворимые рецепторы к VEGF. Дефицит VEGF может лежать в основе системной эндотелиальной дисфункции, почечной ТМА, повреждения подоцитов с распластыванием их ножек и, как следствие, развития АГ, нарушения функции почек и ПУ.
Появились данные, что ПЭ способствует развитию сердечно-сосудистых заболеваний в отдаленном будущем, а рождение маловесных детей сопряжено с развитием терминальной почечной недостаточности спустя многие годы после родов у их матерей. Учитывая, что маловесные дети рождаются, как правило, при развитии ранней ПЭ, вероятно, рано развившаяся ПЭ может рассматриваться и как фактор риска ХБП. Однако, несмотря на высокий интерес нефрологов к проблеме ПЭ в последние годы, данные о влиянии ПЭ на почечное поражение, прогностическом значении сроков развития ПЭ крайне скудны и нуждаются в дальнейшем изучении.


