
Синдром поликистозных яичников (СПКЯ) представляет собой патологический симптомокомплекс, характеризующийся олиго-ановуляцией, гиперандрогенией (клинической и/или биохимической), нарушением структуры и функции яичников. СПКЯ встречается у 8–13% пациенток раннего репродуктивного возраста [1]. Инсулинорезистентность (ИР) встречается среди 35–90% женщин с СПКЯ. Данный показатель в 2–3 раза превышает распространенность ИР при анализе здоровых женщин, стратифицированных по возрасту и индексу массы тела [2]. Также отмечается манифестация метаболического синдрома к 20 годам примерно у 20% девушек [3].
Как известно, для диагностики СПКЯ, по данным Роттердамских критериев, необходимо присутствие двух из трех параметров: клиническая или биохимическая гиперандрогения, овуляторная дисфункция, поликистозная морфология яичников по данным ультразвукового исследования (УЗИ). По международным клиническим рекомендациям 2018 г. у всех пациенток с СПКЯ необходимо оценивать гликемический статус: глюкозотолерантный тест, уровни глюкозы и гликированного гемоглобина в крови в измерениях раз в 2–3 года по крайней мере [4].
Несмотря на многочисленные исследования, посвященные патогенезу СПКЯ, единое понимание не сформировано. В патогенезе СПКЯ принимает участие сочетанное нарушение многочисленных факторов, как генетических, так и эпигенетических [5]. В момент становления начальных этапов эмбриогенеза у плода могут реализовываться дефекты развития, такие как нарушение программирования во время внутриутробного развития метаболической оси у плода, сбой секреторной функции надпочечников. В подростковом возрасте при относительной гиперандрогении и гиперинсулинемии имеют особое влияние характер физической активности и диеты, наличие избыточной массы тела и ожирения, обострение хронических или острых инфекционных заболеваний, что может служить пусковым механизмом нарушения правильного функционирования гипоталамо-гипофизарно-яичниковой и надпочечниковой оси. Также имеют место различные социальные факторы и воздействие окружающей среды, такие как нездоровый образ жизни, курение, злоупотребление алкоголем, переедание, отсутствие физической активности и др. В период беременности особое влияние имеют диета, наличие нейроэндокринных нарушений и осложнений беременности (преэклампсия, соматические патологии, требующие медикаментозной коррекции, угроза прерывания беременности), что может предопределять развитие симптомокомплекса в дальнейшем [6].
Кроме факторов риска развития СПКЯ, особое значение имеют нарушения центральной регуляции синтеза гонадотропных гормонов [7]. В результате влияния многочисленных эндогенных и экзогенных факторов в экстрагипоталамических структурах мозга происходит нарушение обмена нейромедиаторов (нейрокинина В, кисспептина, гамма-аминомасляной кислоты, динорфина, кокаина и амфетамин-регулируемого транскрипта (CART), субстанции Р), возрастает продукция и выделение β-эндорфина и уменьшается синтез допамина, приводя к изменению амплитуды и частоты секреции гонадотропин-рилизинг-гормона (ГнРГ) [8].
Как известно, СПКЯ сопровождается повышенным выбросом лютеинизирующего гормона (ЛГ) и увеличением соотношения ЛГ/ФСГ (фолликулостимулирующий гормон) в связи с увеличенной пульсовой секрецией ГнРГ гипоталамусом. Нейроны в дугообразных ядрах медиобазальных отделов гипоталамуса секретируют различные виды нейротрансмиттеров и нейропептидов, например, нейропептид Y, ГнРГ, агути-подобный пептид, CART, субстанцию Р, дофамин, кисспептин, соматокринин, нейрокинин В, β-эндорфин и соматостатин, которые участвуют в патогенезе СПКЯ. На сегодняшний день активно изучается влияние кисспептина, нейрокинина В и динорфинов в генезе симптомокомплекса. Показано, что данные нейропептиды контролируют выброс ГнРГ через механизм обратной связи, действуя на эстрогеновый рецептор альфа [9]. При взаимодействии кисспептина-нейрокинина В с ГнРГ и антагонистом рецептора нейрокинина 3 наблюдается снижение частоты выброса ЛГ, и, соответственно, уровень сывороточного ЛГ падает. Таким образом, использование антагониста нейрокининового рецептора приводило к уменьшению концентрации тестостерона в сыворотке крови у пациенток с СПКЯ, что рассматривается для применения в клинической практике.
В последние годы в патогенезе СПКЯ большое внимание уделяется сочетанным гормональным нарушениям на фоне метаболических. Как известно, инсулин является одной из причин усиления ЛГ-зависимого синтеза тестостерона; также он способствует подавлению продукции глобулина, связывающего половые гормоны, в печени, тем самым увеличивая содержание биологически активных свободных фракций тестостерона и эстрадиола. Гиперинсулинемия понижает выработку в печени белков, которые связывают инсулиноподобный фактор роста, и, как следствие, приводит к повышению его биодоступности. Chong Feng et al. в исследовании 2018 г. показали, что у пациенток с СПКЯ на фоне ИР происходит уменьшение синтеза глобулина, связывающего половые гормоны, через снижение экспрессии IRS-1, IRS-2, GLUT-4 в PI3K/Akt-опосредованном сигнальном пути, что приводит к усугублению гиперандрогении [10].
Как известно, ожирение, как один из самых часто встречающихся факторов риска развития СПКЯ, является важным его клиническим проявлением. Пациентки с СПКЯ чаще страдают абдоминальным типом ожирения и имеют избыточный вес в 38–88% случаев заболевания [11, 12]. Жировая ткань служит источником большого количества активных биологических веществ (адипокинов, аполипопротеинов). Гипертрофия адипоцитов у пациенток с ожирением и СПКЯ взаимосвязана с локальной гипоксией, инфильтрацией ткани макрофагами, нарушением чувствительности к инсулину [13]. В исследовании Dumesic D.A. et al. показали, что у пациенток СПКЯ репродуктивного возраста, в том числе с нормативными значениями индекса массы тела, в сравнении с группой контроля наблюдалась ИР на уровне жировой ткани и изменение экспрессии ряда генов подкожных абдоминальных жировых стволовых клеток, что коррелировало с гиперандрогенией и метаболическими нарушениями [14]. В перекрестном исследовании у девочек с СПКЯ были показаны пониженная чувствительность к инсулину, уменьшение переработки свободных жирных кислот по сравнению с данными у здоровых девочек, а также дисфункция адипоцитов [15].
Жировая ткань синтезирует энзимы, способные как активировать, так и инактивировать предшественники андрогенов. Фермент альдокеторедуктаза 1-го семейства (AKR1C3) вырабатывается в жировой ткани и способствует образованию тестостерона из андростендиона. Энзим 5α-редуктаза 1-го типа (SRD5A1), которая экспрессируется из жировой ткани, конвертирует тестостерон в дигидротестостерон. Исследования in vivo O'Reilly et al. показали повышение AKR1C3 и SRD5A1 мРНК, что предполагает существование порочного цикла, связывающего биосинтез андрогенов и накопление липидов с ИР и гиперинсулинемией у пациенток с СПКЯ [16, 17].
В исследованиях, посвященных изучению механизмов действия инсулина у пациенток с СПКЯ, использовались клеточные модели фибробластов, адипоцитов и клетки скелетных мышц [18]. При этом исследование инсулинового рецептора не выявило мутаций или структурных нарушений у пациенток с СПКЯ. Авторы сделали вывод, что пострецепторные дефекты на пути передачи сигналов инсулина играют важную роль в этиологии возникновения резистентности к инсулину.
Рецептор инсулина является двудимерным гликопротеином, состоящим из α- и β-субъединиц. Обе субъединицы кодируются одним геном на 19 хромосоме и образуются в результате частичного протеолиза единого предшественника. Взаимодействие инсулина с рецептором приводит к каскаду реакций фосфорилирования. Первично происходит аутофосфорилирование тирозиновых остатков внутриклеточного домена рецептора, что, в свою очередь, приводит к активации рецептора и фосфорилированию остатков серина на субстрате инсулинового рецептора (IRS), способствуя активации внутриклеточных путей дальнейшей передачи сигнала: PI3K (фосфоинозитид-3-киназы) и AKT. После активации IRS способствует закреплению на внутренней мембране гетеродимера фермента фосфатидилинозитол-3-киназы, имеющего в составе регуляторную p85- и каталитическую p110-субъединицы. Далее киназа фосфорилирует мембранный фосфатидилинозитол-4,5-дифосфат до фосфатидилинозитол-3,4,5-трифосфата (PIP3). Существует мнение, что PIP3 играет роль в качестве мембранного якоря для других элементов при воздействии инсулина. После образования PIP3 происходит активация 3-фосфоинозитид-зависимой протеинкиназы-1 (PDK-1). Данная протеинкиназа связывается с ДНК-протеинкиназой (DNA-PK), что, в свою очередь, приводит к двойному фосфорилированию протеинкиназы B (AKT-1). АКТ-1 прикрепляется к мембране под воздействием PIP3, и происходит ее активация. Затем АКТ-1 отсоединяется от мембраны и транслоцируется в цитоплазму и ядро клетки, где способствует фосфорилированию многочисленных белков-мишеней. В ядре активированная протеинкиназа В запускает экспрессию генов гликолиза и липолиза и тормозит экспрессию генов глюконеогенеза и липогенеза. Таким образом, реализуется первый PI3K/Akt сигнальный путь передачи сигнала с инсулина, который вовлечен в метаболические реакции обмена белков, углеводов и липидов, так называемые быстрые и очень быстрые эффекты инсулина (рис. 1) [19].
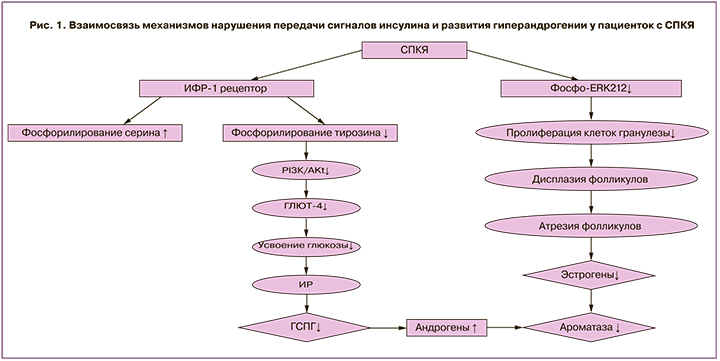
При СПКЯ обнаружено увеличение фосфорилирования серина под влиянием IRS-1, в то время как фосфорилирование тирозина снижено и нарушена активация PI3K. Таким образом, данные нарушения препятствуют передаче сигналов инсулина и способствуют возникновению резистентности к нему, что выражается в недостаточно выраженной активации процессов гликолиза и липолиза, и пролонгированию процессов глюконеогенеза и липогенеза. Существуют данные, сообщающие, что в адипоцитах женщин с СПКЯ аутофосфорилирование инсулиновых рецепторов снижено на треть в сравнении со здоровыми, что коррелирует с резистентностью к инсулину [20].
В организме поглощение глюкозы происходит через ее транспортеры GLUT. Среди них одним из ключевых является тканеспецифический инсулин-регулируемый GLUT4 (SLC2A4), который участвует в гомеостазе глюкозы в физиологических и патологических условиях [21]. Доказано, что уровень GLUT4 снижен в эндометрии у женщин с СПКЯ, страдающих ожирением и гиперинсулинемией [22]. Снижение экспрессии транспортеров глюкозы у пациенток с СПКЯ может быть обусловлено, в том числе, активацией рецептора под влиянием пероксисомных пролифераторов (PPAR –peroxisome proliferator-activated receptors) и фактора транскрипции FOXO1 (forkhead box protein O1). FOXO1 является наиболее распространенной изоформой в инсулинчувствительных тканях поджелудочной железы, печени и жировой ткани. Его функция регулируется AKT, которая, фосфорилируя фактор транскрипции FOXO1, инактивирует его и обуславливает локализацию в цитоплазме, в то время как транскрипция PPAR продолжается [23]. Согласно результатам Zhang et al. (2012), пациентки с СПКЯ характеризуются более высоким уровнем фосфорилирования AKT (p-AKT) в сопоставлении с условно здоровыми женщинами. При этом у пациенток с СПКЯ на фоне ИР выявлена более высокая концентрация p-AKT и активация PI3K/AKT сигнального пути в сравнении с женщинами с СПКЯ без ИР, что обуславливает нарушение процессов захвата GLUT клетками и прогрессирование нарушения толерантности к углеводам [24]. Таким образом, у пациенток с СПКЯ, вне зависимости от наличия избыточной массы тела, в том числе на фоне нормогликемии и нормоинсулинемии, отмечается постпрандиальная гипергликемия, в том числе в связи с нарушением транспорта и утилизации глюкозы [25].
Кроме первого PI3K/Akt сигнального пути быстрых и очень быстрых эффектов инсулина, существует второй механизм передачи сигнала – медленные и очень медленные эффекты инсулина. Данный второй механизм опосредуется сигнальными каскадами митоген-активируемой протеинкиназы (MAPK) и связан с изменением экспрессии ядерных генов, регулирующих рост, пролиферацию и дифференцировку клеток.
Этапы реализации МАРК-сигнального пути включают связывание аутофосфорилированного инсулинового рецептора с белком Shc, являющимся еще одним субстратом инсулинового рецептора. Далее Shc-белок связывается с Grb-белком, что вызывает присоединение его к рецептору. Формирование комплекса Shc-Grb активирует группу GEF-SOS-GAP и способствует фосфорилированию гуанозиндифосфата (ГДФ). Образование ГДФ в составе Ras-белка позволяет активировать протеинкиназу Raf-1, что вызывает ее присоединение к цитоплазматической мембране, дополнительное фосфорилирование по остаткам тирозина, серина и треонина, взаимодействие с инсулиновым рецептором. Активированная Raf-1 фосфорилирует и активирует киназы MAPK-K (MEK) и МАPК (ERK), что изменяет активность белков цитоплазмы. примеру, активация фосфолипазы А2 приводит к образованию арахидоновой кислоты и синтезу эйкозаноидов, активация рибосомальной киназы инициирует трансляцию белков, активация протеинфосфатаз вызывает дефосфорилирование ряда ферментов. Передача сигнала через медленные пути инсулина приводит к активации системного воспаления (увеличивается синтез провоспалительных цитокинов – фактора некроза опухоли (ФНО-α, интерлейкинов (ИЛ)-6, ИЛ-18), изменению синтеза лейкотриенов, а также к активации ферментов гликолиза. Известно, что ФНО-α способствует пролиферации клеток теки, продуцирующих андрогены, стимулирует атрезию фолликулов и гиперандрогению, что выявлено у женщин с СПКЯ [26].
Установлено, что снижение активности PI3K в тека-клетках у пациенток с СПКЯ связано со снижением активности 17-альфа-гидроксилазы. Кроме того, исследования продемонстрировали, что инсулин влияет на синтез гормонов в клетках теки и гранулезы через повышение выработки белка, регулирующего стероидогенез StаR (acute steroidogenic regulatory protein). StaR опосредует транспорт холестерина в митохондрии для синтеза гормонов. Sander et al. обнаружили повышение экспрессии мРНК StAR в клетках гранулезы у женщин с СПКЯ, которые наблюдались в программе ЭКО [27]. CYP11A1 (p450cc) осуществляет первый этап стероидогенеза: преобразование холестерина в прегненолон. Wickenheisser et al. показали, что период полураспада мРНК CYP11A1 (р450сс) увеличен более чем в 2 раза в тека-клетках у женщин с СПКЯ, что приводит к избытку мРНК CYP11A1 при СПКЯ. Liu et al. изучили экспрессию CYP11A1 в фолликулах в ранней и поздней стадиях развития у пациенток с СПКЯ и без после лапароскопической резекции яичников [17]. Выявлен более высокий уровень мРНК CYP11A1 в фолликулах на ранней стадии развития у женщин с СПКЯ, что связано с нарушением фолликулогенеза и формированием фенотипа поликистозных яичников.
CYP17A1 является членом суперсемейства цитохромов Р450 и участвует в метаболизме и синтезе стероидов, в результате которого производятся прогестины, минералокортикоиды, глюкокортикоиды, андрогены и эстрогены, преобразует прегненолон и прогестерон в их 17-альфа-гидроксилированные продукты, через 17-гидроксилазный и 17,20-лиазный активные центры синтезируется дегидроэпиандростерон (ДГЭА) и андростендион [28]. Заключающим ферментом в синтезе стероидов является 17-гидроксистероид-дегидрогеназа, которая участвует в преобразовании эстрона в эстрадиол, ДГЭА в андростендион, андростендиона в тестостерон, дегидротестостерона в 5-альфа-андростендион [29]. В клетках теки у пациенток с СПКЯ отмечается повышение синтеза андрогенов в связи с активацией 17α-гидроксилазы и 17,20-лиазы. У пациенток с СПКЯ при сопоставлении со здоровыми установлена повышенная активность не только CYP17A1, но и CYP11A1, и ароматазы CYP19A1, что приводило к увеличению продукции прогестерона, 17-ОН-прогестерона и тестостерона (рис. 2) [17].
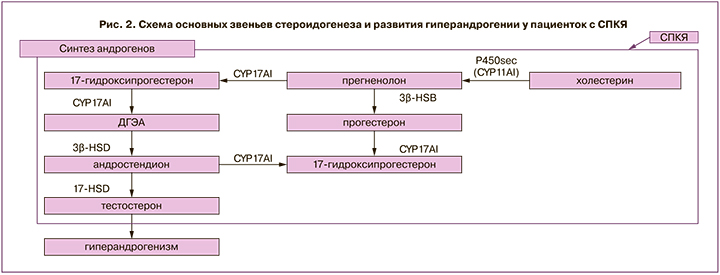
Андрогены имеют разнонаправленное действие, что было продемонстрировано на животной модели у обезьян. Было показано, что андрогены усиливают действие ФСГ в мелких антральных фолликулах, но оказывают ингибирующее действие на более крупные фолликулы [30]. С одной стороны, андрогены конвертируются в эстрогены, способствующие росту фолликулов, с другой стороны, избыточный синтез андрогенов нарушает фолликулогенез и селекцию доминантного фолликула. Считается, что из-за относительного дефицита ФСГ и эстрадиола, а также повышения по механизму обратной связи синтеза ЛГ и экспрессии рецепторов ЛГ происходит атрезия фолликулов [31]. Морфология поликистозных яичников представлена преобладанием мелких антральных фолликулов от 2 до 5 мм и избыточным развитием клеток теки [32].
Кроме того, показано, что инсулин или инсулиноподобный фактор роста-1 может стимулировать синтез фактора роста эндотелия сосудов (VEGF-A) в лютеинизированных клетках гранулезы. VEGF является основным регулятором физиологического ангиогенеза. Уровни полного овариального васкуляризационного индекса (total ovarian vascularization index (VI)) и индекса васкуляризации (VFI – vascularization flow index) по данным УЗИ и допплерометрии у женщин с СПКЯ выше, чем в группе контроля [33]. Повышение уровня васкуляризации может опосредовать аномальный рост клеток теки интерна, приводя к активации стероидогенеза и гиперандрогении. Было предположено, что повышение секреции VEGF и увеличение ангиогенеза может быть одним из звеньев патогенеза СПКЯ [34].
Строма яичника обеспечивает структурную основу, подвергающуюся динамическим изменениям для поддержки роста фолликула. Строма яичников у женщин с СПКЯ отличается большей ригидностью, чем в группе здоровых женщин [26]. Показано, что строма яичников при СПКЯ представлена фибробластами, имеющими веретенообразную форму с ядрами удлиненной формы, при этом клетки тесно прилежат друг к другу и не имеют четких границ. Иногда в строме яичника встречаются участки более часто расположенных ядер, которые, по-видимому, свидетельствуют об очаговой клеточной гиперплазии. Среди таких клеток нередко можно встретить фигуры митоза. Клеточные элементы межуточной ткани у пациенток с СПКЯ могут подвергаться лютеинизации, образуя очаги текоматоза [34].
У пациенток с СПКЯ уровень конечных продуктов гликирования (КПГ, AGP, advanced glycation end products) как в системном кровотоке, так и в тканях яичника значительно выше, чем у здоровых женщин, что обусловлено активацией оксидативного стресса и митохондриальной дисфункции. КПГ образуются в результате химических реакций между аминогруппами белков и карбонильными группами углеводов. Предполагаемая роль КПГ в изменении стероидогенеза при СПКЯ до конца не изучена. Diamanti-Kandarakis et al. показали, что сывороточные уровни КПГ и экспрессия рецептора к КПГ (RAGE, receptor for AGP) у пациенток с СПКЯ в несколько раз выше при сравнении с условно здоровыми женщинами, включая женщин с СПКЯ с ИР без гипергликемии, а также отмечена связь с уровнем тестостерона [35]. Депонирование КПГ в клетках теки и гранулезы яичников и чрезмерно повышенная экспрессия рецепторов к КПГ стимулирует НАДФ-оксидазу и основной воспалительный сигнальный путь фактора NF-kB, что повышает продукцию активных форм кислорода и синтез провоспалительных факторов (эндотелина-1, ИЛ-1, ИЛ-6, ИЛ-8, ФНО-α). В результате повышение уровня КПГ в системном кровотоке и тканях яичника на фоне снижения антиоксидантной защиты при СПКЯ ведет к пролонгированию воспалительного и оксидативного стресса, нарастанию эндотелиальной дисфункции в дальнейшем [36].
Как известно, митохондрии отвечают за поддержание клеточного гомеостаза и являются уникальными органеллами, которые имеют свой собственный геном мтДНК и отвечают за синтез клеточной энергии, детоксикацию, регулируют высвобождение вторичных мессенджеров и клеточную сигнализацию, синтез стероидных гормонов и гема, клеточную пролиферацию и дифференцировку, аутофагию и апоптоз. Накапливаются данные, что при СПКЯ ИР, гиперандрогения и нарушения фолликулогенеза могут быть опосредованы митохондриальной дисфункцией и оксидативным стрессом [34]. В исследовании показано, что изменение чувствительности к инсулину в скелетных мышцах у женщин с СПКЯ ассоциировано со снижением экспрессии генов митохондриального метаболизма [37]. У взрослых пациенток с СПКЯ выявлено нарушение митохондриального катаболизма интермедиатов цикла Кребса, увеличение активности гликолиза при снижении глюконеогенеза в печени. К повреждающим проявлениям оксидативного стресса относят активацию перекисного окисления липидов, окислительную модификацию белков и ДНК, инактивирование ферментов [38]. У пациенток с СПКЯ выявлена более высокая активность глутатионпероксидазы, супероксиддисмутазы, каталазы на фоне потребления антиоксидантов неферментативного звена, таких как восстановленный глутатион, что взаимосвязано с метаболическими изменениями при заболевании [39].
Третьим основным сигнальным путем инсулина, задействованным в патогенезе СПКЯ, является mTORC-1 и -2. Белок mTOR является серин-треониновой протеинкиназой, участвующей в регуляции роста и пролиферации клеток, подвижности и выживании, а также влияет на синтез белков и транскрипцию. В передачу сигналов с участием mTOR-пути интегрированы различные сигнальные каскады. Блокада mTOR приводит к торможению сигнала к пролиферации и остановке клеточного цикла в фазе G1 [40].
Известно, что mTOR является каталитической субъединицей в двух различных молекулярных комплексах [41]. Он активируется инсулином, ростовыми факторами, фосфорной кислотой, аминокислотами (в основном лейцином) и оксидативным стрессом. Основной функцией mTORC1 является инициация трансляции различных генов и синтеза белков, в том числе в связи с активацией биогенеза рибосом. Также mTORC1 способствует торможению аутофагии. Известно, что пониженный уровень питательных веществ и ростовых факторов, куркумин, кофеин, рапамицин оказывают ингибирующее действие на mTORC1, в то время как mTORC2 регулирует организацию цитоскелета и выживание клеток [42].
Группа исследователей Roa et al. в 2009 г. выявили, что mTOR является ключевым звеном в развитии половой зрелости и активации секреции ЛГ. mTOR-путь может быть активирован лейцином, что изменяет секрецию ЛГ у мышей в периоде пубертата через систему кисспептина (Kiss-1 gene) и его рецептор (Kiss-1R). В исследовании были описаны рецепторы к лейцину в нейронах кисспептина, которые принимают участие в метаболической регуляции Kiss-1 [42]. Молекулярные механизмы передачи действия лейцина на транскрипцию гена Kiss-1 и его потенциальную интеграцию с другими регуляторами экспрессии Kiss-1 еще недостаточно изучены [43]. Инактивация путей mTOR в центральной нервной системе приводит к снижению секреции Kiss-1 и к умеренному снижению мРНК ГнРГ в гипоталамусе, что приводит к уменьшению концентрации ЛГ в гипофизе [44]. Подавление сигнального пути mTOR рапамицином было связано с угнетением гонадотропной оси в период полового созревания, снижением уровней эстрадиола и ЛГ, что вызывало гипоплазию матки и яичников. Группа исследователей Yaba и Demir в 2012 г. отметили важную роль mTORC1 и mTORC2 в развитии СПКЯ. Результаты работ показали, что активация mTOR-путей может регулировать рост фолликулов in vivo, в частности, инициируя рост и пролиферацию фолликулярных клеток яичника в мышиной модели СПКЯ [45]. При ингибировании mTOR рапамицином уменьшаются рост фолликулов in vitro и пролиферация гранулезных клеток [46]. Allemand et al. выявили, что повышенное фосфорилирование S6K и mTOR приводит к ингибированию ISR-1 и резистентности к инсулину. Полученные данные на мышиной модели свидетельствуют, что активация пути mTORC1 может принимать участие в развитии ИР в скелетных мышцах мышей и гиперинсулинемии у пациенток с СПКЯ [47].
Заключение
Таким образом, ИР, гиперинсулинемия и гиперандрогения развиваются по нескольким возможным механизмам. В данном обзоре представлены основные сигнальные пути, участвующие в патогенезе развития СПКЯ как самостоятельно, так и при взаимодействии друг с другом. У пациенток с выявленной ИР при СПКЯ установлена более высокая концентрация p-AKT и активация быстрого PI3K/AKT сигнального пути в сравнении с женщинами с СПКЯ без ИР, что обуславливает нарушение процессов захвата GLUT клетками и прогрессирование нарушения толерантности к углеводам. Повышение уровня андрогенов способствует снижению активности фермента ароматазы в клетках гранулезы, ингибируя конвертирование андрогенов в эстрогены, что участвует в прогрессировании атрезии фолликулов. Второй сигнальный путь, MARK-ERK1/2, активирован у пациенток с СПКЯ меньше, чем у здоровых женщин, что препятствует пролиферации клеток гранулезы, и также связан с усилением атрезии фолликулов. Кроме того, отмечается роль третьего сигнального пути, mTOR, в развитии нарушений фолликулогенеза при СПКЯ. Помимо описанных механизмов, на сегодняшний день активно изучается влияние нейропептидов, таких как кисспептин, нейрокинин В и динорфины, на генез симптомокомплекса СПКЯ. На основании полученных данных о вовлеченных сигнальных путях в развитие симптомокомплекса подтверждена взаимосвязь порочного круга ИР и гиперандрогении. Фундаментальное понимание механизмов развития ИР при СПКЯ особенно актуально с точки зрения возможности разработки действенных лечебных схем ведения пациенток с раннего репродуктивного возраста в дальнейшем.


