
Преждевременная недостаточность яичников (ПНЯ) – мультифакторная патология, в этиопатогенезе которой существенная роль принадлежит генетическим нарушениям [1, 2]. Моногенные формы ПНЯ наследуются в соответствии с законом Менделя. При этом, в большинстве случаев имеет место аутосомно- рецессивный тип наследования с низким риском повторения у потомства. В настоящее время известно более 20 моногенных форм ПНЯ, зарегистрированных в международной медицинской базе наследственных заболеваний OMIM (Online Mendelian Inheritance in Man, менделевское наследование у человека), среди которых ведущая роль принадлежит гену FMR1 [3]. Известно, что наибольшее значение для прогноза потомства представляют Х-сцепленные рецессивные формы синдрома ПНЯ. Ген FMR1 (fragile mental retardation) – ген умственной отсталости картирован на длинном плече Х хромосомы в локусе Хq27.3. В 5’ нетранслируемой области его 1-го экзона содержатся тринуклеотидные CGG повторы [4, 5].
Ген FMR1 экспрессируется в ооците, в гранулезных и лютеиновых клетках, а также в нейронах [5]. Недостаточность выработки белка FMRP (fragile X mental retardation protein) вследствие мутации в гене FMR1 лежит в основе патологии фолликулогенеза, приводя к ановуляции. При этом, в ЦНС наблюдается расстройство нейрогенеза с исходом в когнитивные нарушения и умственную отсталость. Экспансия тринуклеотидных CGG-повторов (более 200) сопровождается аномальным метилированием и отсутствием экспрессии гена FMR1 [6]. Вышеуказанная нестабильность тринуклеотидов может приводить к формированию нескольких генетических синдромов – преждевременная недостаточность яичников I типа (Premature Ovarian Failure 1), синдром Мартина–Белл (FRAXA, Х-сцепленная умственная отсталость) и синдром тремора и атаксии (Fragile X associated tremor/ataxia syndrome, FXTAS).
Согласно российским и международным рекомендациям, всем женщинам с ПНЯ и их родственницам, в настоящее время следует определять CGG повторы в гене FMR1 [7 ,8].
В практике гинекологов в Российской Федерации за норму принят средний диапазон распределения CGG повторов от 28 до 36 тринуклеотидов [9]. Согласно данным Американского колледжа медицинской генетики (ACMG), рассматриваются три варианта нарушений числа CGG повторов в гене FMR1 – «истинная» мутация (синдром ломкой Х-хромосомы, число повторов более 200), премутация гена FMR1 (55–199 тринуклеотидов) и их значения в пределах «серой зоны» (45–54 повторов) [10]. Помимо вышеуказанной классификации, по данным Gleicher N. et al., аллели менее 28 тринуклеотидов расцениваются как «короткие повторы», в то время как диапазон в пределах 37–44 – как удлинение CGG повторов [11].
Частота встречаемости премутации в гене FMR1 имеет выраженные половые различия, у женщин 1:180–1:259, в то время как, у мужчин 1:230– 1:810 случаев [12]. При семейной преждевременной недостаточности яичников ее распространенность составляет 11,5%, а при спорадической форме – 3,2% [10, 13]. Впервые в 1994 г. Schwartz C.E. et al. в многоцентровом исследовании продемонстрировали, что у носительниц премутации в гене FMR1 менопауза наступала в возрасте до 40 лет в три раза чаще, чем в группе контроля [14]. В 2004 г. обнаружено, что у больных с увеличением длины триплетных повторов в гене FMR1, вплоть до премутации гена, выявлено статистически значимое повышение уровня фолликулостимулирующего гормона (ФСГ), снижение уровней ингибина А и В, не соответствующие их возрастной норме в сочетании со снижением овариального резерва. При этом, только в 16–25% случаев у носительниц премутации гена FMR1 развивается ПНЯ [15]. На протяжении последних десяти лет Gleicher N. et al. также доказано формирование ПНЯ как при коротких триплетных повторах гена FMR1, так и у носительниц тринуклеотидов в пределах «серой зоны» [11]. Таким образом, на сегодняшний день в качестве биомаркера прогнозирования преждевременного старения яичников, возможно, использовать наличие числовых нарушений CGG повторов в гене FMR1.
В доступной литературе крайне мало исследований, посвященных анализу особенностей формирования репродуктивного, соматического и неврологического статуса у детей, рожденных от матерей с ПНЯ носительниц аномального числа в гене FMR1.
Цель исследования: оценка особенностей здоровья детей, рожденных от матерей с ПНЯ с учетом наследования аномального числа CGG повторов в гене FMR1.
Материалы и методы
Проспективное одноцентровое исследование серии непоследовательных случаев проведено на базе отделения гинекологической эндокринологии и института репродуктивной генетики ФГБУ «НМИЦ АГП им. В.И. Кулакова» Минздрава России. В исследование были включены 90 женщин с ПНЯ, проходившие обследование и/или лечение, в соответствии с нашей клинической деятельностью. Набор пациентов проводился с 2018 по 2021 гг. Диагноз ПНЯ был установлен на основании Российских клинических рекомендаций [8]. Исследование одобрено локальным этическим комитетом ФГБУ «НМИЦ АГП им. В.И. Кулакова» Минздрава России, регистрационный номер 5 от 14.04.2016 г. От каждой пациентки было получено письменное информированное согласие на участие в исследовании в соответствии с Хельсинкской декларацией, из них 19 пациенток подписали согласие на обследование детей.
Критерии включения в исследование: установленный диагноз ПНЯ; возраст женщин на момент постановки диагноза от 18 до 39 лет; кариотип 46 ХХ.
Критерии исключения из исследования: отсутствие детей у женщин с ПНЯ.
Критерии невключения из исследования: наличие первичной или ятрогенной гипер-, нормо- и гипогонадотропной аменореи.
Для верификации диагноза ПНЯ оценивалось функциональное состояние гипоталамо-гипофизарно-яичниковой системы на основании определения уровней фолликулостимулирующего гормона (ФСГ), эстрадиола (Е2), в сыворотке крови электрохемилюминесцентным методом на автоматическом иммунохимическом анализаторе Cobas е411 (Roche Diagnostics GmbH, Германия). Антимюллеров гормон (АМГ) определяли в сыворотке крови количественно методом ферментно-усиленного двухступенчатого иммуноанализа сэндвич-типа с помощью набора AMH Gen II ELISE (Beckman Coulter, США).
Ультразвуковое исследование органов малого таза проводилось на аппарате 2000 Toshiba SSA-240 (Япония) трансвагинальным конвексным датчиком частотой 7,5 МГц.
Кариотип исследовали по общепринятому методу Seabright (G-окраска).
Методика определения числа повторов CGG в гене FMR1 с помощью амплификации промоторной области гена, включающей CGG-область, была выполнена методом ПЦР с использованием флуоресцентно меченых праймеров и определения точной длины ПЦР-продуктов фрагментным анализом на генетическом анализаторе серии 31хх Applied Biosystems (набор реактивов «ДНК-Технология») в соответствии с протоколом «Стандартные операционные процедуры (СОП) по определению количества повторов CGG в гене FMR1».
Результаты
В исследование вошли 90 женщин с ПНЯ, их средний возраст составил 33,5 лет. Средний возраст дебюта заболевания – 31 год. Средние значения гормонального обследования 90 женщин с ПНЯ составили для ФСГ – 61,88 мМЕд/мл, Е2 – 44,7 пмоль/л, АМГ – 0,13 нг/мл.
На первом этапе работы всем женщинам с установленным диагнозом ПНЯ было проведено определение CGG повторов в гене FMR1 (табл. 1).

У 45/90 пациенток (50%) выявлялись различные числовые нарушения тринуклеотидов по отношению к нормативным показателям. При этом, у 8/90 (8,9%) женщин выявлена премутация в гене, у 4/90 (4,4%) – тринуклеотидные повторы находились в пределах «серой зоны» и у 33/90 (36,7%) пациенток обнаружены короткие повторы. В двух случаях у носительниц премутации в гене FMR1 детей не было, в связи с чем, в дальнейшем, они были исключены из исследования. На втором этапе исследования проводилось генетическое тестирование 27 детей, рожденных от 19 носительниц аллелей с аномальным числом CGG повторов.
В первой серии случаев анализировались варианты наследования потомством аномальных тринуклеотидных повторов от матерей с премутацией в гене FMR1 и их репродуктивный статус. Результаты FMR1-тестирования матерей-носительниц премутации и их потомства представлены в таблице 2. У 6 носительниц премутационных аллелей менархе было своевременным и до дебюта заболевания регулярный ритм менструации, а также сохраненная детородная функция, что подтверждалось наступлением 18 беременностей, 12 из которых закончились рождением 7 мальчиков и 5 девочек; в остальных 6 случаях было произведено искусственное прерывание беременностей по желанию женщин. Детородная функция у женщин, входящих в данную серию случаев была реализована до 30 лет (в среднем 27,6 лет). Средний возраст дебюта заболевания составил 32 года. Первый аллель у матерей находился в диапазоне от 20 до 30, второй – от 80 до 86 триплетных повторов.
При анализе наследования числовых нарушений гена FMR1 у детей, рожденных от носительниц премутации выявлено следующее их распределение: у сына (1/12 детей, 8,3%), выявлены триплетные повторы в пределах нормальных значений за счет наследования первого материнского аллеля. У 3 дочерей (3/12, 25%) наблюдались короткие повторы, при этом, первый аллель они унаследовали от матери, а второй, вероятно, от отцов. При детальном изучении анамнеза выявлено, 2 девочки находились в препубертатном периоде, у третьей – менархе было своевременным и до настоящего времени регулярный ритм менструации. У 2 девочек и 4 мальчиков (6/12, 50%) обнаружено наследование от матерей премутации в гене FMR1. Число триплетных CGG повторов колебалось от 78 до 106, при этом, их увеличение в процессе наследования наблюдалось у 4 детей, у 1 – сокращение и в одном случае – стабильная передача. При детальном изучении анамнеза выявлено, что у 1 мальчика половое созревание в соответствии с возрастом. Трое мальчиков находились в препубертатном периоде, дополнительно у одного из них были выявлены числовые хромосомные аномалии по типу синдрома Клайнфельтера. У 2 дочерей отмечен тяжелый яичниковый фенотип ПНЯ с очень ранним дебютом заболевания в 17 и 18 лет (через 4–5 лет после менархе). Обращает на себя внимание, что у их матерей дебют заболевания наступил значительно позже (через 19 и 21 год после менархе). У 2 сыновей (2/12, 16,7%) была обнаружена экспансия тринуклеотидов до полной мутации гена FMR1, родились мальчики с синдромом Мартина–Белл с типичными клиническими (психопатические и речевые нарушения в виде двигательной расторможенности, признаки аутизма) и фенотипическими проявлениями заболевания (долихоцефалия, выступающий лоб, удлиненное лицо, массивный подбородок, крупные ушные раковины, эпикант, крупные кисти и стопы, гипермобильность суставов, макроорхизм).
Во второй серии случаев анализировались варианты наследования потомством аномальных тринуклеотидных повторов от матерей с тринуклеотидами в пределах «серой зоны» в гене FMR1 и их репродуктивный статус. Результаты генетического обследования матерей с повторами в пределах «серой зоны» и их потомства представлены в таблице 3. У всех обследованных нами пациенток носительниц триплетных аллелей в пределах «серой зоны» менархе было своевременным, до дебюта заболевания ритм менструаций регулярный и сохраненная детородная функция, что подтверждалось наступлением 7 беременностей, 4 из которых закончились рождением 2 мальчиков и 2 девочек. Средний возраст на момент родов – 25,5 лет. Изучение числа триплетных повторов у детей выявило следующее их распределение, в 50% (2/4) в пределах популяционной нормы, в 50% (2/4) – наследование, коротких тринуклеотидных повторов. Все дети, рожденные в данной серии случаев, находились в препубертатном периоде без диагностированных неврологических и психических нарушений.
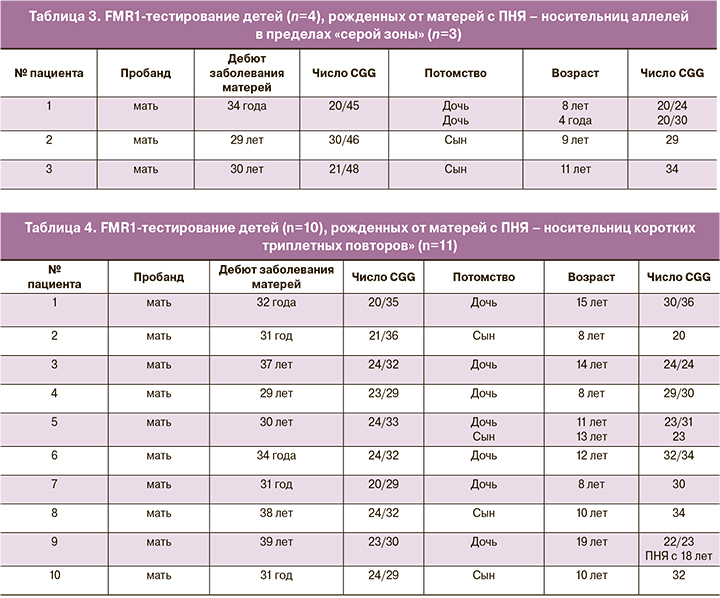
В третьей серии случаев анализировались варианты наследования потомством аномальных тринуклеотидных повторов от матерей с короткими повторами в гене FMR1 и их репродуктивный статус. Результаты генетического обследования матерей с короткими повторами в гене FMR1 и их потомства представлены в таблице 4. У всех обследованных нами пациенток носительниц триплетных аллелей в пределах укороченных повторов менархе было своевременным, до дебюта заболевания ритм менструаций регулярный и сохраненная детородная функция, что подтверждалось наступлением 17 беременностей 11 из которых закончились рождением 4 мальчиков и 7 девочек, остальные беременности закончились искусственным их прерыванием по желанию женщин. Средний возраст пациенток на момент родов составил 29,7 лет. У матерей первый аллель попадал в диапазон от 20 до 24 CGG повторов, второй – от 29 до 36 тринуклеотидов. Стабильная передача коротких повторов была выявлена у 3 дочерей и 2 сыновей (5/11, 45,5%), у 4 девочек и 2 мальчиков (6/11, 54,5%) триплетные повторы были в пределах рефересных значений. Из 11 детей 7 находятся в препубертатном периоде, у 3 – половое созревание в соответствии с возрастом. Все дети без диагностированной соматической и неврологической патологии. У 1 дочери 19 лет (1/11, 9,1 %) был диагностирован тяжелый фенотип ПНЯ в возрасте 18 лет, в то время как у матери, дебют заболевания отмечен в 31 год.
Обсуждение
На протяжении многих лет носительство премутации в гене FMR1 не связывали с развитием каких-либо соматических заболеваний. Однако, в последние десятилетия доказано, что у носительниц премутации возможно развитие коморбидной патологии, в структуре которой в 16–25% случаев встречается ПНЯ. При этом, помимо нарушений репродуктивной системы могут развиваться аутоиммунная патология, Х-ассоциированный тремор-атаксический синдром, а также нейрокогнитивные и эмоционально-личностные расстройства. В связи с вышеизложенным, обсуждение полученных нами результатов по оценке особенностей здоровья детей, рожденных от матерей с ПНЯ носительниц аномального числа тринуклеотидных CGG повторов в гене FMR1, представляет собой определенную как научную, так и практическую значимость.
По данным многочисленных литературных источников, доказано, что риск экспансии CGG повторов в последующем поколении до состояния полной мутации возрастает по мере увеличения у матерей числа тринуклеотидов в гене FMR1. Согласно данным Nolin S.L. et al., при обследовании 936 женщин с различными числовыми повторами в гене FMR1 в рамках премутационного диапазона был рассчитан риск формирования в последующем поколении полной мутации с учетом длин материнских повторов. При диапазоне CGG тринуклеотидов в пределах 55–59 (n=26) он составил 3,7%, в интервале числовых повторов 80–89 (n=133) – 57,8%, при 90–99 тринуклеотидов (n=118) риск достигал 80%. Максимальный риск (94–100%) наблюдался при длине CGG повторов от 100 до 109 (n=26). Вышеуказанное исследование, к сожалению, было проведено без учета овариального резерва женщин и развития в последующем у них ПНЯ [16]. По нашим данным, у матерей с ПНЯ с триплетными повторами в рамках премутации 22/80 и 20/86 экспансия CGG тринуклеотидов с исходом в полную мутацию и рождением мальчиков с синдромом Мартина–Белл в течение одного поколения выявлена в 16,7% случаев, что в 3,7 раза ниже при сопоставимых числовых повторах в вышеуказанной работе [16] (рисунок).

В то же время, частота формирования синдрома Мартина–Белл в нашем исследовании соотносится с результатами работы Beke A. et al., в которой показано, что у 9 женщин с ПНЯ при носительстве премутации в гене FMR1, синдром Мартина–Белл у потомства был диагностирован еще реже, в 11,1% случаев [17].
В конце ХХ в. было выявлено, что в норме тринуклеотидные CGG повторы в гене FMR1 прерываются AGG тринуклеотидами, чаще всего в положениях 10 и 20, что обеспечивает стабильность их передачи. В случае отсутствия AGG прерываний или их недостаточного числа триплетные CGG повторы становятся крайне нестабильными с возможностью выраженной экспансии и формирования синдрома Мартина–Белл в последующем поколении [18]. Однако, к сожалению, расширенный анализ гена FMR1 c одновременным определением CGG и AGG тринуклеотидов матерям с премутацией в гене FMR1 нами выполнен не был.
В ходе нашей работе в 50% случаев (6/12) мы наблюдали стабильное наследование детьми премутации в гене FMR1 от женщин с ПНЯ. У 2 дочерей с премутацией отмечался тяжелый фенотип ПНЯ, с дебютом заболевания в 17 и 18 лет, в то время как в работе Табеевой и соавт. при обследовании 200 пациенток с ПНЯ средний возраст дебюта заболевания составил 29,4±7,66 лет [19].
В последние годы в литературе активно обсуждается роль коротких CGG повторов в гене FMR1, при которых наблюдаются расстройства репродуктивной и нервной систем [11]. В нашей работе в 36,7% случаев выявлены носительницы коротких повторов. В 2014 г. Mailick M.R. et al. было представлено уникальное долгосрочное исследование, включающее 10 317 человек, за которыми наблюдали в течение 54 лет. К концу исследования возраст респондентов достиг 72 лет и на финальном этапе работы 3469 (51,5%) женщинам было проведено определение СGG повторов в гене FMR1, в ходе которого выявлено 20,8% носительниц коротких повторов, что в 1,8 раз реже в сравнении с нашими пациентками с ПНЯ. Авторы отметили, что у женщин, носительниц коротких повторов в гене FMR1 в 2 раза чаще диагностировался рак молочной железы и в 4 раза – рак эндометрия, чем у носительниц нормальных СGG повторов. У их потомства значительно чаще диагностировалась тяжелая умственная отсталость в сравнении с популяционными данными (отношение шансов 1,68, р<0,05). В то время как у мужчин носителей коротких повторов преобладал тремор-атаксический синдром [20]. По нашим данным, при стабильной передаче коротких CGG повторов в 9,1% случаев, у дочери выявлен тяжелый фенотип ПНЯ с дебютом заболевания в 18 лет.
Известно, что триплетные повторы в пределах «серой зоны» подвержены экспансии в последующих поколениях, однако, в нашем исследовании, ввиду крайне ограниченной выборки, данный феномен подтвержден не был [21, 22].
В результате проведенного исследования доказано, что для детей, рожденных от матерей с различными вариантами аномальных CGG тринуклеотидов в гене FMR1 характерны следующие варианты наследования числовых повторов:
1) нормальные триплетные CGG повторы обнаружены в 33,3% (9/27) случаев;
2) короткие повторы встречались в 37% (10/27). При стабильной передаче коротких повторов в 9,1% случае выявлен тяжелый фенотип ПНЯ у дочери.
3) премутации гена FMR1 обнаружена в 22,2% (6/27) с формированием у девочек тяжелого фенотипа ПНЯ.
4) расширение повторов до полной мутации с рождением мальчиков с синдромом Мартина–Белл выявлено в 7,5% (2/27).
В настоящее время развитие молекулярно-генетических технологий позволяет пациенткам-носительницам премутации и гетерозиготной мутации в гене FMR1 планировать рождение биологически родного ребенка. При обсуждении репродуктивного прогноза им принято рекомендовать медико-генетическое консультирование, при котором пациентка с ПНЯ получает информацию о возрастных ограничениях детородного потенциала (желательна реализация беременности до 30 лет), о высоком риске рождения мальчика с синдромом Мартина–Белл и возможностях преимплантационного генетического тестирования эмбрионов (определение пола эмбриона и селекция его в пользу женского) [23]. Также пациентки должны быть информированы, что при спонтанном наступлении беременности им показано проведение пренатальной (дородовой) диагностики, которая может включать: определение пола плода с 10 недель беременности путем проведения неинвазивного пренатального скрининга ДНК плода (НИПС) по крови матери; ультразвуковую диагностику врожденных аномалий и половой принадлежности плода; инвазивную пренатальную диагностику кариотипа (в том числе, пола) и мутаций в гене FMR1 у плода путем биопсии хориона в 10–13 недель, или амниоцентеза в 16–20 недель беременности.
Однако, селекция эмбриона/плода по половой принадлежности в пользу женского пола, не всегда позволяет избежать появления на свет девочки с репродуктивными проблемами, унаследованными от матери. Ресурсом для уточнения характера наследования тринуклеотидных повторов может быть более детальное исследование влияния локусов прерывания AGG [24]. Требует дальнейшего исследования неоднозначная роль коротких повторов в развитии ПНЯ и рисках для потомства. Учитывая наличие множества других моногенных форм ПНЯ пациенткам «серой зоны» и с нетипичными проявлениями, может быть рекомендован поиск мутаций в других генах, ассоциированных с ПНЯ, методом полноэкзомного секвенирования.
Ограничения исследования. Небольшая выборка пациентов. Отсутствие результатов тестирования СGG повторов в гене FMR1 отцов. Отсутствие результатов исследования на локусы AGG вкраплений.
Заключение
Неблагоприятный исход для потомства наблюдается в серии случаев от матерей носительниц премутационных аллелей в гене FMR1, что традиционно давало основания для рекомендаций таким женщинам достижения беременности за счет донорских яйцеклеток или эмбрионов в программе вспомогательных репродуктивных технологии. Таким образом, в случае планирования беременности у этой категории пациенток клинический генетик должен учитывать точное количество CGG повторов в гене FMR1, а также AGG прерываний, чтобы получить надежные результаты для индивидуальной оценки риска.
По нашему мнению, необходимо проведение генеалогического анализа пациенток с ПНЯ носительниц аномального числа CGG повторов в гене FMR1 c учетом генетического тестирования их родственников 1–2 степени родства для определения вариантов наследования. Для обнаружения на раннем этапе фенотипических особенностей у потомства целесообразно проведение коллегиального консультирования, включая педиатра, детского гинеколога, генетика и невролога с целью необходимости назначения детям поведенческой или медикаментозной терапии.


