
Распространенность метаболического синдрома (МС) продолжает неуклонно расти во всем мире; по данным ряда эпидемиологических исследований, она варьирует от 12,4 до 28,5% у мужчин и от 10,7 до 40,5% у женщин. Особое внимание заслуживает рост распространенности МС среди женщин репродуктивного возраста, среди которых данная патология диагностируется в 25–30% случаев. Известно, что пациенты с ожирением и МС имеют дополнительный риск развития заболеваний сердечно-сосудистой системы, а также особенно высокий риск тромбоэмболических осложнений [1]. На сегодняшний день крайне важным является изучение патогенетической взаимосвязи МС и риска развития тромбоэмболических осложнений. Результаты исследований демонстрируют ассоциацию МС с гиперкоагуляцией, при этом отмечается повышение активности плазматического звена гемостаза, снижение фибринолиза, эндотелиальная дисфункция, повышение активности тромбоцитов [2]. Инсулинорезистентность, являющаяся основным патогенетическим фактором МС, может обусловливать развитие данного процесса посредством угнетения синтеза и высвобождения как оксида азота, так и простациклина в эндотелии, а также повышения синтеза и биодоступности эндотелина-1. Оксид азота, помимо вазодилятации, ингибирует адгезию и агрегацию тромбоцитов, снижает проницаемость сосудистой стенки и ингибирует пролиферацию сосудистых гладкомышечных клеток [3, 4].
Глюкозотоксичность, липотоксичность, хронический провоспалительный статус тоже играют роль в развитии эндотелиальной дисфункции, а также усиливают инсулинорезистентность, способствуя развитию дальнейших метаболический нарушений [5].
По данным ряда исследований, при ожирении и МС отмечено повешенное содержание макрофагов в жировой ткани; нарушение взаимодействия между макрофагами и адипоцитами является основной причиной развития дисфункции последних. Избыток жировой массы повышает уровень внутриклеточных липидов и приводит к гипертрофии и гиперплазии адипоцитов [6–8]. Образование большого количества адипоцитов ведет к повышенному содержанию свободных жирных кислот, обладающих проатерогенным эффектом на клетки эндотелия, макрофаги и клетки гладкой мускулатуры сосудов и ингибирующих антилиполитическую активность инсулина [9]. Свободные жирные кислоты также связывают неспецифический толл-подобный рецептор-4 макрофагов, активируют ядерный фактор κВ и, следовательно, увеличивают продукцию фактора некроза опухоли-ɑ (ФНО-ɑ). Продукция макрофагами ФНО-ɑ в дальнейшем индуцирует липолиз адипоцитов и увеличивает концентрацию молекул адгезии на поверхности адипоцитов и моноцитов, привлекает моноциты в жировую ткань и способствует их дифференциации в макрофаги. Таким образом, увеличение продукции ФНО-ɑ макрофагами приводит к образованию паракринного порочного круга, ведущего к развитию хронического провоспалительного статуса [10].
Повышенный уровень свободных жирных кислот оказывает патологическое воздействие и на сосудистую стенку, вызывая апоптоз эндотелиальных клеток, индуцирующий экспрессию ФНО-ɑ, интерлейкина (ИЛ)-8, молекул клеточной адгезии и, как следствие, привлечение моноцитов в эндотелий [11].
В результате данных процессов происходит пролиферация гладкомышечных клеток сосудистой стенки и синтез компонентов внеклеточного матрикса, способствующий задержке в интиме сосуда липопротеинов низкой плотности (ЛПНП) и формированию атеросклеротической бляшки [12].
Чрезмерное накопление липидов в клетке вызывает эндоплазматический и митохондриальный стресс, проявляющийся в нехватке эндоплазматического протеина, формировании жировых капель, повышении уровня лактата, разобщении окислительных процессов в митохондриях и продукции реактивных форм кислорода и свободных радикалов. Таким образом, наблюдаемое при ожирении внутриклеточное повреждение адипоцитов трансформируется путем воздействия повышенного уровня ФНО-ɑ, адипокинов и медиаторов воспаления в системный воспалительный ответ [7].
Адипонектин, основной протеин, секретруемый адипоцитами, обладает антиатеросклеротическим и противовоспалительным действием [13]. Адипонектин ингибирует продукцию ФНО-ɑ и ИЛ-6 адипоцитами и эндотелиальными клетками, снижает экспрессию молекул адгезии на поверхности этих клеток, блокирует накопление липидов и трансформацию макрофагов в пенистые клетки, ингибирует обусловленный липопротеидами низкой плотности местный оксидативный стресс и пролиферацию эндотелия, снижает образование свободных радикалов и стимулирует синтез эндотелием оксида азота [14]. Адипонектин также обладает и антитромботическим действием. В исследованиях на мышах было доказано, что повышенная экспрессия адипонектина замедляет тромбообразование [15]. Наблюдающаяся при ожирении перестройка местных нейроэндокринных механизмов и повышенный оксидативный стресс приводят к снижению продукции протеинов жировой тканью, в том числе адипонектина [16]. Таким образом, снижение уровня адипонектина приводит к увеличению воспаления, привлечению в очаг воспалительных клеток и росту атеросклеротических бляшек [17].
Помимо адипокинов адипоцитами осуществляется синтез целого ряда воспалительных цитокинов, таких как ФНО-ɑ, ИЛ-1, ИЛ-6, матриксные металлопротеиназы (MMP), способные разрушать белки внеклеточного матрикса, и их ингибиторы (TIMP). По последним данным, жировая ткань продуцирует до 30% цитокинов данного типа. ИЛ-6 повышает поверхностную адгезию эндотелиальных клеток, снижает продукцию адипонектина и активность липопротеинлипазы, увеличивая поглощение липидов макрофагами и их трансформацию в пенистые клетки, что в итоге приводит к формированию атеросклеротической бляшки [18].
Другим важным патогенетическим фактором атеротромбогенеза при ожирении и МС является гиперактивность тромбоцитов и снижение их чувствительности к влиянию антиагрегантов. Известно, что у пациентов с МС, особенно при наличии нарушенной толерантности к углеводам и с абдоминальным ожирением, отмечается повышение агрегации и адгезивной способности тромбоцитов, как спонтанной, так и стимулированной [9, 19–21]. На сегодняшний день имеется ряд объяснений данному факту: нарушение аффинности и/или снижение числа гликопротеиновых рецепторов к адгезивным протеинам на поверхности тромбоцитов; повышение активности фибриногена, нарушение метаболизма и структуры мембран тромбоцитов, а также изменения в интратромбоцитарных сигнальных путях [20, 22, 23]. Нарушения в структуре мембран тромбоцитов могут являться основной причиной их гиперчувствительности и гиперфункции при МС, а также негативно влиять на ряд метаболических процессов, например, повышать мобилизацию ионов кальция, а также синтез и высвобождение тромбоксана [24]. Было выявлено, что наличие эндотелиальной дисфункции и дислипидемии, особенно гипертриглицеридемии, может способствовать усилению агрегации тромбоцитов, способствуя повышению риска развития тромбозов [20, 24–26].
Имеется ряд данных, указывающих на ассоциацию гипертриглициридемии, характерной для МС и гиперкоагуляции. В частности, повышение уровня липопротеинов очень низкой плотности (ЛПОНП) и остаточных липопротеинов, наблюдаемое при МС, может усиливать активность тромбоцитов и активировать коагуляцию, способствуя формированию протромбиназных комплексов [20, 23–25]. ЛПОНП также могут активировать экспрессию гена ингибитора активатора плазминогена-1 (PAI-1), приводя к увеличению концентрации и активности последнего в плазме крови, что ассоциировано с повышенной агрегацией тромбоцитов и тромбо-образованием [20, 24, 25, 27]. Также было показано, что число мембранных микропузырьков тромбоцитов, способствующих коагуляции посредствам выделения анионных фосфолипидов и тканевых факторов, ассоциировано с количеством компонентов МС [28].
Ряд клинических исследований показал увеличенную активность комплексов «тканевой фактор (TF) – фактор коагуляции VII» и опосредованную данным комплексом гиперкоагуляцию у пациентов с ожирением [14]. Также у таких пациентов была выявлена повышенная экпрессия мРНК тканевого фактора в адипоцитах и клетках адвентиции окружающей сосуды [6]. Повышенный уровень TF, «комплексов TF – фактор коагуляции VII», фрагментов протромбина, обусловливающий протромботический статус у пациентов с ожирением, прямо пропорционален индексу массы тела (ИМТ) и подвергается редукции при снижении веса [15]. Кроме того, было доказано, что лептин увеличивает экспрессию TF в периферических мононуклеарных клетках, подтверждая связь гиперлептинемии и протромботического статуса у пациентов с ожирением [29].
Повышенный уровень других компонентов коагуляционного каскада, таких как фибриноген, фактор коагуляции VIII, комплексы тромбин-антитромбин III, фактор фон Виллебранда, также наблюдается при ожирении. Считается, что адипокины и цитокины, секретированные гипертрофированными и гиперплазированными адипоцитами, приводят к нарушению печеночного метаболизма и увеличению секреции факторов коагуляции [30]. В том числе возрастает и уровень фибриногена, ввиду его повышенного синтеза печенью в условиях индуцированного ИЛ-6 воспалительного статуса, характерного для пациентов с ожирением. Уровень фибриногена, фактора коагуляции VIII, белка острой фазы также пропорционален ИМТ и подвержен снижению при потере веса [31].
Большое число клинических исследований подтверждают повышенный уровень PAI-1 и снижение фибринолитической активности у пациентов с ожирением, в особенности с абдоминальной формой. Считается, что повышенный уровень гормонов и цитокинов (ФНО-ɑ, ИЛ-1, ИЛ-6, ангиотензин-2 и др.), наблюдающийся при ожирении, нарушает аутокринную и паракринную регуляцию жировой ткани и приводит к повышенному синтезу PAI-1. Наибольшей секреторной активностью и продукцией PAI-1 обладают гипертрофированные адипоциты абдоминальной зоны. Была выявлена прямая корреляция между уровнем PAI-1, тканевого активатора плазминогена (tPA), фибриногеном и ИМТ. Снижение массы тела у пациентов с ожирением приводило к нормализации показателей фибринолитической системы [32].
Другим модулятором фибринолиза, уровень которого также повышается при ожирении, является ингибитор фибринолиза активируемый тромбином (TAFI). TAFI удаляет аргининовые и лизиновые остатки с С-терминального конца фибрина, участвующего в связывании с tPA и плазминогеном, и, таким образом, ослабляет фибринолиз [24].
Комбинация повышенного уровня TAFI и PAI-1 при ожирении обусловливает наблюдающийся у данных пациентов гипофибринолитический фенотип и их склонность к гиперкоагуляции.
В последние годы активно изучается роль генетически обусловленного гипофибринолиза у пациентов с МС. По данным ряда авторов, у женщин с МС абсолютное большинство среди генетических форм тромбофилии составили наследственные дефекты, предрасполагающие к гипофибринолизу (полиморфизм генов PAI-1 4G/4G, фибриногена 455 G/A, ангиотензин-превращающего фермента I/D, тканевого активатора плазминогена) [33].
Следует отметить, что в свою очередь у беременных с МС высокий уровень PAI-1 не только влияет на процесс имплантации плодного яйца, повышает риск ранних и поздних самопроизвольных выкидышей, развития тяжелых форм гестоза, но также является независимым фактором тромбофилии, повышающим риск тромботических осложнений на протяжении всего гестационного процесса [34]. При этом сочетание МС, генетических дефектов гемостаза, гипергомоцистеинемии, циркуляции антифосфолипидных антител значительно увеличивает эти риски [35, 36]. Согласно данным ряда отечественных авторов, у большинства пациенток с МС именно тромбофилия является сложным интегральным фактором, занимающим ведущее место в патогенезе развития акушерских и тромботических осложнений [33, 37].
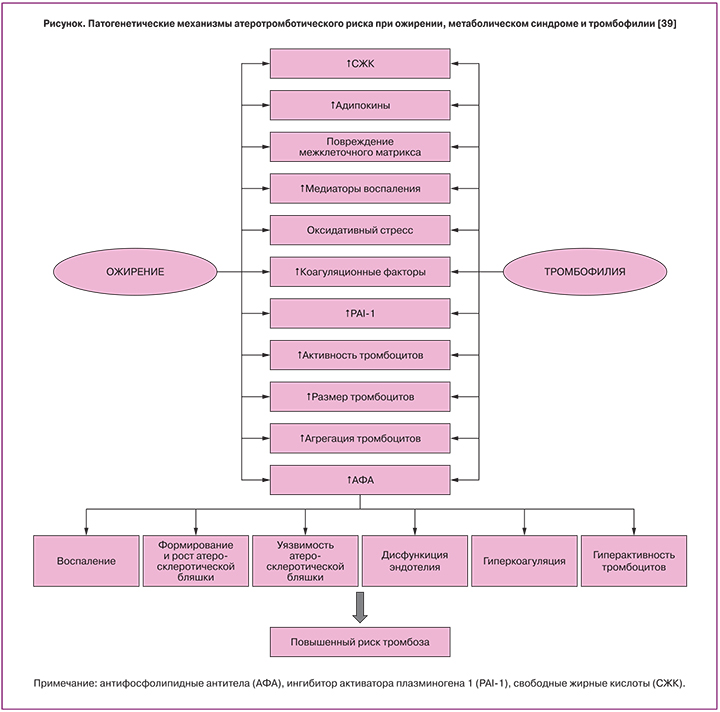
По данным ряда исследований, скрытые генетические тромбофилии и циркуляция антифосфолипидных антител у пациентов с МС являются одним из пусковых механизмов повреждения эндотелия, нарушения регуляции тонуса сосудистой стенки, увеличения тромбогенного потенциала, влияют на состояние микроциркуляции и способствуют развитию эндотелиопатии [21]. При эндотелиопатии снижается выработка эндотелиоцитами естественных антикоагулянтов и оксида азота и усиливается высвобождение индукторов активации тромбогенеза, способствующих образованию тромбина, фибрина, тромбоцитарных агрегатов. Развиваются процессы микро- и макротромбирования сосудов [5, 38].
Неоднократно была описана связь ожирения и тромбоэмболических эпизодов, причинами развития которой являются характерные для ожирения провоспалительный и протромботический статусы, способствующие дисфункции эндотелия и развитию атеросклероза [5, 39]. Локальный воспалительный процесс в интиме сосудов у пациентов с ожирением возникает в результате длительного воздействия высоких уровней холестерина и ЛПНП, частицы которых проникают в интиму сосудов, связываются с протеингликанами, что впоследствии приводит к модификации эндотелиальных и гладких мышечных клеток сосудов. Модифицированные клетки начинают продуцировать провоспалительные молекулы и цитокины. Данная провоспалительная среда привлекает и трансформирует прибывшие в очаг воспаления моноциты в макрофаги. Макрофаги проникают в интиму сосудов, поглощают и накапливают проникшие туда липидные частицы, и, в конечном итоге, превращаются в пенистые клетки. Накопление воспалительных клеток и макрофагов с липидными частицами в интиме сосудов вызывает синтез протеинов экстрацеллюлярного матрикса, приводит к образованию фиброзной капсулы и способствует дальнейшему накоплению липидов, что, в свою очередь, приводит к некрозу макрофагов и клеток гладкой мускулатуры сосудов и разрушению сосудистой стенки [40]. Увеличение липидно-некротического ядра приводит к истончению фиброзной капсулы и повышает уязвимость атеросклеротической бляшки. Если данный процесс прогрессирует, то происходит разрыв фиброзной капсулы, и компоненты липидно-некротического ядра попадают в кровеносное русло и приводят к образованию тромбов в просветах сосудов. В конечном итоге атеротромботический процесс приводит к манифестации тромбоэмболического эпизода [41].
В последнее время появляется все больше эпидемиологических и клинических исследований, свидетельствующих о взаимосвязи ожирения и венозной тромбоэмболии [39]. По данным ряда научных работ, риск венозной тромбоэмболии увеличивается вдвое у лиц с ИМТ более 30 кг/м², данная взаимосвязь значительно усиливается с увеличением ИМТ. Таким образом, патогенетической основой увеличения риска венозной тромбоэмболии у пациентов с ожирением и МС является избыток жировой ткани, который наряду с такими факторами, как провоспалительный статус, оксидативный стресс, патология генов гемостаза, нарушение синтеза факторов коагуляции, циркуляция антифосфолипидных антител может также значительно увеличивать риск артериальных тромбозов и случаев повторной тромбоэмболии. МС ассоциирован с венозной тромбоэмболией, более того, отдельные компоненты данного синдрома, являющиеся следствием ожирения, демонстрируют наиболее сильную патогенетическую связь с тромбозами [28].
Вопрос о назначении противотромботической терапии должен быть рассмотрен в отношении пациентов с диагностированным венозным тромбозом или имеющих тромбоз в анамнезе, а также при наличии таких независимых факторов риска тромбоэмболии, как тромбофилия, ожирение, МС, особенно если они сочетаются с беременностью или неудачными исходами беременности в анамнезе. Отдельного внимания заслуживает рассмотрение проблемы назначения противотромботической терапии при беременности. Следует отметить, что крайне важно перед зачатием либо в ранние сроки беременности выявлять женщин, которым, возможно, потребуется назначение антикоагулянтной терапии. Рекомендуется проводить профилактику тромбозов всем пациенткам с МС и наследственной тромбофилией. При этом особое внимание заслуживают пациентки с МС и диагностированной мультифакторной формой тромбофилии [35, 42].
Препаратами выбора как до, так и во время беременности являются производные гепарина. Как гепарин, так и низкомолекулярные гепарины не проникают через плацентарный барьер и признаны безопасными. По данным рандомизированного исследования с применением низкомолекулярных гепаринов данная терапия позволяет также улучшить исходы беременности при наличии в анамнезе гестационных осложнений, обусловленных патологией плаценты, независимо от наличия тромбофилии [43].
Имеются данные, указывающие, что низкомолекулярные гепарины могут обладать не только противотромботическим, но и противовоспалительным свойством. Противовоспалительная активность низкомолекулярных гепаринов может быть обусловлена их способностью ингибировать прокоагулянтную активность лейкоцитов и моноцитов, снижать синтез провоспалительных цитокинов клетками – индукторами воспаления, подавлять экспрессию молекул адгезии, подавлять экспрессию TF активированным эндотелием за счет стимулирования высвобождения ингибитора пути тканевого фактора (TFPI), или липопротеин-ассоциированного ингибитора коагуляции (LACI-фактор) и антикомплементарным действием [44].
Также имеется небольшое количество научных работ, рассматривающих снижение избыточной массы тела как фактор профилактики риска венозной тромбоэмболии у лиц с ожирением и МС [1].
Заключение
Таким образом, ассоциация МС и его компонентов с тромботическими осложнениями не вызывает сомнений, а лежащие в ее основе патогенетические механизмы (рисунок), обусловленные эндокринными и паракринными нарушениями, провоспалительным и протромботическим статусами, требуют назначения своевременной и грамотной противотромботической терапии. В отдельную группу особого риска следует отнести беременных с МС, имеющих повышенный риск развития тромбоэмболических осложнений на протяжение всего гестационного процесса и послеродового периода и нуждающихся в проведении специфической противотромботической терапии, безопасной во время беременности и в период кормления грудью [45].
И несмотря на значимое количество исследований, посвященных изучению факторов риска и патогенетических механизмов развития тромбоэмболических осложнений у пациентов с МС, проблема выявления причин и способов коррекции нарушений окончательно не решена и требует дальнейшего изучения. При этом ежегодно увеличивающийся процент лиц с ожирением и МС среди женщин репродуктивного возраста ставит новые задачи по оптимизации их подготовки к беременности, проведению профилактики тромбогеморрагических осложнений во время беременности, родоразрешению и в послеродовом периоде.


