
Миома матки (лейомиома) представляет собой доброкачественную моноклональную опухоль миометрия – мышечного слоя матки. Такие опухоли диагностируются у 40–50% женщин репродуктивного возраста, чаще в позднем репродуктивном и пременопаузальном периодах [1, 2]. Однако в последние десятилетия случаи диагностирования миом у женщин до 30 лет участились. В настоящее время диагноз «миома матки» встречается у 3,3–7,8% женщин до 30 лет, причем заметную долю таких пациенток составляют женщины с нереализованной репродуктивной функцией [3].
Симптомы, вызываемые миомами, зависят от расположения и величины опухоли; чаще всего это боли, кровотечения и нарушение функции соседних органов вследствие сдавливания их разрастающейся опухолью. Современные методы лечения включают в себя гормонотерапию и хирургические методы – выполняемые лапароскопическим, лапаротомическим и влагалищным доступами миомэктомии, а нередко и гистерэктомии (что особенно нежелательно в репродуктивном возрасте женщины). Ни один из методов лечения миомы матки не лишен недостатков и не может исключать осложнения и рецидивы. Развитие миомы может вести к бесплодию, осложнять течение беременности, а также вызывать проблемы при вынашивании и в родах [4]. Все это делает миому матки одной из важнейших угроз репродуктивному здоровью женщин.
До недавнего времени не существовало единого мнения о причинах развития миомы матки. Обычно выделяли несколько факторов, способствующих развитию миомы матки: нарушение выработки половых гормонов; хронические воспалительные заболевания женской половой сферы (хронический сальпингоофорит, инфекции, передающиеся половым путем); аборты, внутриматочные контрацептивы; заболевания эндокринных желез (щитовидной железы, надпочечников и др.). Среди этих факторов также обычно фигурирует «генетическая предрасположенность к возникновению миомы матки», что подтверждается наличием «семейных форм» данного заболевания у 5–10% больных [5], однако конкретных механизмов такой предрасположенности до недавнего времени описано не было [6, 7].
 Поиск генетических причин развития миом ведется уже более 20 лет. Поскольку миомы являются моноклональными опухолями, то есть развиваются из одной клетки-предшественницы, наибольшие усилия исследователей были сосредоточены на соматических изменениях генома опухолей. Еще в 90-х годах прошлого века с помощью FISH были описаны крупные перестройки в геномах миом – такие как трисомия 12 и делеция на 7-й хромосоме (del(7)(q22q32), размер которой может достигать нескольких миллионов нуклеотидов [8, 9], а также целый ряд других крупных дупликаций и делеций [10–12]. По мере развития методов детекции геномных перестроек были обнаружены также соматические делеции и дупликации меньшего размера, характерные для генома миом [12, 13]. Это, в первую очередь, транслокации, затрагивающие гены семейства HMGA (например t(12;14)(q15;q23-24) и t(6;14)(p21;q23-24), а также делеции на хромосомах 1, 22 и др. [14].
Поиск генетических причин развития миом ведется уже более 20 лет. Поскольку миомы являются моноклональными опухолями, то есть развиваются из одной клетки-предшественницы, наибольшие усилия исследователей были сосредоточены на соматических изменениях генома опухолей. Еще в 90-х годах прошлого века с помощью FISH были описаны крупные перестройки в геномах миом – такие как трисомия 12 и делеция на 7-й хромосоме (del(7)(q22q32), размер которой может достигать нескольких миллионов нуклеотидов [8, 9], а также целый ряд других крупных дупликаций и делеций [10–12]. По мере развития методов детекции геномных перестроек были обнаружены также соматические делеции и дупликации меньшего размера, характерные для генома миом [12, 13]. Это, в первую очередь, транслокации, затрагивающие гены семейства HMGA (например t(12;14)(q15;q23-24) и t(6;14)(p21;q23-24), а также делеции на хромосомах 1, 22 и др. [14].
Наиболее значимым открытием последних лет в области патогенеза миом стало описание соматических мутаций в гене MED12, который кодирует одну из субъединиц медиаторного комплекса РНК-полимеразы II. На протяжении последних 5 лет был опубликован целый ряд работ, в которых показано, что соматические мутации в экзоне 2 гена MED12 встречаются в миомах с частотой 50–70% [15–18]. Как правило, речь идет об однонуклеотидных заменах в кодонах 43 и 44.
В нашей стране до настоящего времени не проводилось системных исследований соматических мутаций в гене MED12 в тканях миом. В единственной публикации [19] отмечается, что в миомах, удаленных у российских женщин, встречаются мутации в данном гене, аналогичные мутациям, обнаруженным в США и странах Европы.
Целью нашей работы было определить спектр и частоты различных соматических мутаций в тканях миом в последовательности экзона 2 гена MED12, которые характерны для российской популяции.
Материал и методы исследования
В 2015 году в отделении оперативной гинекологии ФГБУ НЦАГиП были собраны образцы ткани 50 миом от 42 пациенток (от 1 до 4 миоматозных узлов от каждой), а также аликвоты крови всех пациенток. Сбор образцов тканей миом производился непосредственно во время миомэктомии. Фрагменты тканей помещались в физиологический раствор, отправлялись в биобанк ФГБУ НЦАГиП им. В.И. Кулакова, и замораживались на -70°С для последующего хранения в коллекции. Выделение ДНК проводили набором Qiagen (США). Амплификацию ДНК проводили на приборе ДТпрайм (ООО «ДНК-Технология», Россия). Полимеразная цепная реакция (ПЦР) проводилась с праймерами на участок экзона 2 гена MED12 (общая длина фрагмента 320 н.п.). Последовательности фрагментов были определены путем секвенирования методом Сенгера на приборе ABI PRISM 3130 (Applied BioSystems, США).
Для работы с последовательностями использовался редактор BioEdit (Hall). Достоверность двойного сигнала на хроматограмме проверялась как на прямом, так и на обратном прочтении. Только в случае такого двойного подтверждения мутация признавалась достоверной. В случае детектирования мутации в экзоне 2 гена MED12 проводилось выделение ДНК из крови пациентки и последующий анализ на мутации в экзоне 2 гена MED12. Отсутствие мутации в геномной ДНК служило основанием для подтверждения того факта, что обнаруженные в ДНК миомы мутации являются соматическими.
Результаты исследования
Выборка и данные по пациенткам
Средний возраст пациенток, вошедших в исследованную выборку, составил 38,3 года (25–49 лет). Среди них множественные миомы (2 узла и более) были выявлены у 23 пациенток, у остальных 19 имелись крупные одиночные опухоли. При этом размеры узлов варьировали от мелких (1,5 см и менее в диаметре) до самых крупных, диаметр которых достигал 20 см. Для крупных одиночных миом среднее значение диаметра составило 9,6 см.
Однонуклеотидные замены в экзоне 2 гена MED12
Анализ полученной выборки последовательностей показал, что различные варианты соматических мутаций в экзоне 2 гена MED12 были обнаружены в 25 из 50 образцов тканей миом. Доля опухолей, в которых были детектированы мутации, составила 50% образцов, тогда как доля пациенток, в миомах которых были обнаружены мутации, составила 43%. Данные о типах и частотах выявленных мутаций приведены в табл. 1.
Всего в экзоне 2 гена MED12 удалось выявить 6 вариантов однонуклеотидных замен в 3 позициях. Так, три варианта однонуклеотидных мутаций были выявлены в позиции 131 (G>А в семи миомах, G>С в трех и G>Т в двух миомах). При этом мутация 131 G>А является наиболее частой и по литературным данным [7]. Замены в позиции 130G>Т и 130G>С были обнаружена только в одиночных образцах от двух разных пациенток, тогда как замена 107 Т>С – в двух миоматозных узлах одной и той же пациентки. Эта замена является уникальной, в литературных источниках и базах данных она отсутствует.
Во всех случаях обнаружения мутаций были проверены образцы крови пациенток. Все обнаруженные мутации являлись соматическими.
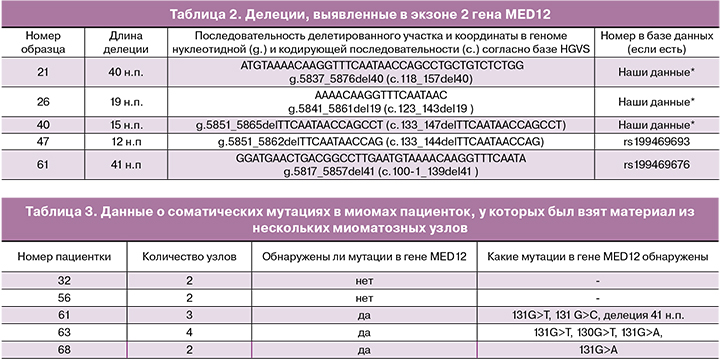
Делеции
Помимо однонуклеотидных замен достаточно частыми, но мало представленными в публикациях вариантами соматических мутаций в экзоне 2 гена MED12, являются делеции. Ранее разными авторами были описаны единичные случаи обнаружения таких делеций; часть из них занесены в базу данных dbSNP (www.ncbi.nlm.nih.gov/snp/). В исследованной нами выборке было выявлено 5 делеций, две из которых соответствовали уже зарегистрированным в базе данных, а 3 описаны нами впервые. Длины и нуклеотидные последовательности всех обнаруженных делеций представлены в табл. 2.
Особенности множественных миом матки
В исследованную выборку были включены образцы тканей нескольких (2 и более) миоматозных узлов, взятых у одних и тех же пациенток, оперированных по поводу множественного миоматоза. Всего таких пациенток было пять. Данные представлены в табл. 3.
У двух пациенток не было найдено никаких мутаций в гене MED12. В двух других случаях множественной миомы у первой пациентки было исследовано 4 миоматозных узла, в первом детектирована замена 131 G>А, во втором – 131 G>Т, в третьем замен не было обнаружено, а в четвертом найдена замена 130 G>Т. У другой пациентки было исследовано 3 узла, при этом в двух обнаружены замены 131 G>С и 131 G>Т, а в третьем – делеция длиной 41 нуклеотид. У пятой пациентки в одном узле была выявлена мутация 131G>A, тогда как во втором никаких мутаций обнаружено не было.
Обсуждение
Основной особенностью гена MED12, изучению соматических мутаций в котором посвящена данная работа, является тот факт, что практически все обнаруженные в нем соматические мутации локализованы в экзоне 2. Это касается как однонуклеотидных замен, так и делеций. В ранее опубликованных работах было показано, что именно участок, кодируемый экзоном 2, в белке субчастицы 12 медиаторного комплекса РНК-полимеразы II отвечает за взаимодействие с циклином С. Любая замена или делеция, затрагивающая кодоны 43 и 44, нарушает это взаимодействие и ведет к нарушению работы клеточного каскада, отвечающего за регуляцию работы РНК-полимеразы II [20]. Однако какие конкретные изменения происходят в клетке при нарушении взаимодействия белка MED12 и циклина С, и каким образом это может вести к превращению нормальной клетки миометрия в клетку-предшественницу миомы, пока остается невыясненным.
В недавно опубликованном исследовании [21] было показано, что встраивание в геном мышей мутантного аллеля гена MED12 (самой распространенной мутации, 131G>A) ведет к развитию миом из тех клеток, где работает только мутантный аллель. При этом в случае полной инактивации гена MED12 в некоторых клетках мезенхимы матки мышей не работало ни одной копии гена, то есть не наблюдалось его экспрессии. Однако никаких изменений в тканях миометрия у таких мышей обнаружено не было. На основании этих результатов авторами был сделан вывод, что полная утрата функции продукта гена MED12 не вызывает развития миом. Когда же исследователям удалось получить мышей, в некоторых клетках миометрия которых экспрессировался только мутантный вариант гена MED12, то у таких мышей начиная с 8-й недели развития наблюдались патологические изменения миометрия, гистологически сходные с лейомиомами.
Все приведенные выше данные позволяют считать ген MED12 одним из ключевых генов, связанных с патогенезом миом.
Полученные нами данные подтвердили распространенность соматических мутаций в гене MED12. Хотя полученные нами частоты и оказались несколько меньше данных по другим популяциям, несомненно, что и в российской популяции довольно высока частота соматических мутаций в гене MED12 (около 50% по нашим данным и около 70% в ранее опубликованной работе [19]).
Особого внимания и дальнейшего изучения заслуживают обнаруженные нами делеции, поскольку ранее опубликованные данные подтверждают, что для развития миомы необходим функционирующий белок – продукт гена MED12. Однако большая часть делеций, в настоящее время выявленных в экзоне 2, приводит к сдвигу рамки считывания, что должно нарушать трансляцию мРНК и биосинтез белка MED12.
В целом, современные исследования демонстрируют наличие как минимум трех «молекулярных типов», то есть различных путей соматических перестроек генома, ведущих к развитию миом [12, 17, 22]. Помимо мутаций в гене MED12, описаны также различные виды соматических хромосомных перестроек, которые характерны для миом, наиболее частая из которых – транслокация 12q14 [11, 12], затрагивающая гены семейства HMGA. Как правило, такие транслокации наблюдаются в миомах, в которых нет соматических замен в гене MED12. Крупные хромосомные перестройки, характерные для миом, согласно литературным данным, а также нашим предварительным результатам, также чаще всего встречаются в миомах без мутаций в гене MED12.
Заключение
Таким образом, современный подход к разработке методов ранней оценки риска развития лейомиом заключается в составлении «молекулярного портрета» каждой опухоли для того, чтобы отнести ее к тому или иному типу. Тщательный сбор клинических данных и сопоставление динамики развития опухолей с «молекулярным портретом» опухоли, возможно, будет иметь ценность в ранней диагностике сложных случаев развития множественных миом.
Основная задача дальнейших исследований – изучение молекулярных механизмов формирования соматических мутаций (в первую очередь наследственных факторов, обусловливающих повышенную частоту возникновения таких мутаций) и поиск взаимосвязей между типом мутации и прогнозом течения заболевания, а также риском рецидивирования после хирургического удаления миом, то есть «агрессивностью» течения заболевания, что очень важно для практического здравоохранения. Ранее нами [5] были опубликованы данные о рецидивном течении миом у больных с «семейными формами» данного заболевания, которые подтверждают существование наследственных факторов, повышающих риск развития миом. Поиск таких факторов является в настоящее время одним из важнейших направлений исследований в области изучения молекулярно-генетических механизмов развития миомы матки.


