
 Введение
Введение
Синдром Блоха-Сульцбергера (синдром недержания пигмента – СНП) – это редкое мультисистемное генетическое заболевание, доминантное Х-сцепленное, поражающее кожу и другие органы, развивающиеся из нейроэктодермы, – волосяные луковицы, зубы, ногти, центральную нервную систему, иммунную систему. Частота встречаемости составляет 1,2:100 000 населения, болеют девочки, так как плоды мужского пола погибают внутриутробно [1–3]. СНП обусловлен мутацией в гене IKBKG, известном как NEMO (эссенциальный модулятор ядерного фактора-каппа-B), кодирующем регуляторный белок, названный IKK (NEMO), расположенный на хромосоме Xq28, который активирует гены, участвующие в выживании клеток, воспалении и иммунитете [5]. В 65% случаев мутации происходят de novo. Клетки, лишенные IKK-гамма, являются мишенями для апоптоза, вызванного фактором некроза опухоли. Клиническая картина неспецифична, так как нет четкой корреляции «генотип–фенотип». Заболевание начинается в периоде новорожденности, характерные кожные поражения являются первым проявлением, проходят четыре стадии: везикуло-буллезную в неонатальном периоде, веррукозную, гиперпигментаци, гипопигментации и атрофии. В старшем возрасте, помимо типичных кожных проявлений, могут быть челюстно-лицевые аномалии, офтальмологические нарушения, патология ЦНС, аномалии придатков кожи, молочных желез. Пациенты, у которых нет клинически значимого поражения ЦНС и глаз, имеют хороший прогноз.
Клинический случай
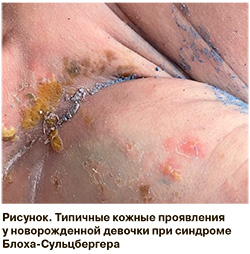
Девочка родилась в одном из регионов РФ. Матери 25 лет, анамнез не отягощен. Беременность первая, во II триместре отмечались герпетические высыпания на губах, на лице. Роды на 37-й неделе, масса тела – 2920 г, длина – 51 см, окружность головы – 33 см, оценка по шкале Апгар – 8/8 баллов. При рождении отмечалась обильная сыпь: везикулы с мутным содержимым на неизмененной поверхности кожи, более выраженные в области живота, верхних и нижних конечностей, в паховой области, на наружных половых органах (рисунок). С подозрением на неонатальную инфекцию кожных покровов ребенок был изолирован и в первые сутки жизни переведен в отделение патологии новорожденных детской областной больницы, где была заподозрена врожденная герпетическая инфекция. При обследовании вирус простого герпеса не выявлен, однако был назначен курс терапии ацикловиром, а также антибактериальная терапия (линезолид, нетимилцин), так как с кожи был высеян эпидермальный стафилококк, чувствительный к линезолиду. Кожу обрабатывали водными и спиртовыми растворами анилиновых красителей, банеоцином. Высыпания подсыхали, превращались в корочки, к двум неделям жизни кожа практически очистилась, однако на 17-е сутки опять появились обильные свежие высыпания по всей поверхности кожи, после чего коллеги обратились за телемедицинской консультацией в федеральный центр. При консультации, учитывая характер высыпаний, их расположение и динамику, был диагностирован синдром Блоха-Сульцбергера и рекомендован перевод в Центр, так как важно не только уточнить диагноз, но и оценить тяжесть состояния, что определяется поражением других органов и систем. При поступлении на 19-е сутки жизни на коже туловища, верхних и нижних конечностей, в паховой области отмечались линейно расположенные везикуло-буллезные элементы желтого цвета, местами окрашенные анилиновыми красителями, а также плотные инфильтрированные розовые папулы. Были отменены все антибактериальные, противовирусные, противогрибковые препараты. Консультации специалистов, всестороннее обследование позволили подтвердить диагноз, а также патологические признаки со стороны других органов и систем. Проводилась только симптоматическая наружная терапия. Учитывая различную морфологию высыпаний на разных стадиях, дифференциальная диагностика проводилась с бактериальной инфекцией, а также с инфекцией, вызванной вирусом простого герпеса, с неонатальной ветряной оспой, невусом сальных желез, буллезным эпидермолизом. При проведении молекулярно-генетического исследования патогенные варианты, имеющие отношение к фенотипу пациента, не были обнаружены, однако известно, что при проведении целевого мутационного анализа ДНК, выделенной из периферической крови, распространенная делеция в IKBKG присутствует в 70–80 % случаев [6]. Диагноз ставится клинически на основании основных критериев (типичные элементы сыпи, распределение высыпаний по линиям Блашко, стадийность кожных проявлений) и вспомогательных (челюстно-лицевые аномалии, офтальмологические нарушения, патология ЦНС, аномалии придатков кожи и молочных желез). Везикулярные поражения на ранней стадии обычно требуют только бережного ухода за кожей с использованием подсушивающих, нейтральных моющих и смягчающих средств, использование глюкокортикоидных препаратов противопоказано. После проведения полного обследования и обучения мамы уходу ребенок был выписан домой.
Заключение
В неонатальном периоде характерные кожные проявления синдрома недержания пигмента необходимо дифференцировать с инфекционно-воспалительными заболеваниями кожи, требующими принципиально другой терапии. Несмотря на то что это редкое заболевание, диагноз ставится клинически, что позволяет избежать полипрагмазии, прежде всего необоснованного назначения антибактериальной терапии.
Литература:
1) Swinney C.C., Han D.P., Karth P.A. Incontinentia Pigmenti: A Comprehensive Review and Update. Ophthalmic Surg Lasers Imaging Retina. 2015;46:650.
2) Landy S.J., Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993;30:53.
3) Narayanan M.J., Rangasamy S., Narayanan V. Incontinentia pigmenti (Bloch-Sulzberger syndrome). Handb Clin Neurol. 2015;132:271.
4) Fusco F., Pescatore A., Steffann J., et al. Clinical utility gene card: for incontinentia pigmenti. Eur J Hum Genet. 2019;27:1894.
5) Scheuerle A.E., Ursini M.V. Incontinentia Pigmenti. 1999 Jun 8 [Updated 2017 Dec 21]. In: Adam M.P., Ardinger H.H., Pagon R.A., et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Bookshelf URL: https://www.ncbi.nlm.nih.gov/books/


